Genetics 101
Genetics is a vast and increasingly complicated field of science. The information here will help to provide a basic background in general genetics concepts and can be a resource as you come up with questions. For more information, explore our library of articles and downloadable resources under the Learn and Resources tabs.
A visit with a genetic counselor can help address your questions and concerns about genetic health, including genetic testing options during pregnancy, cancer risks, and the role genetics play in shaping your health. GSF’s team of certified and experienced genetic counselors can help you understand and address these and other concerns.
The Basics of Genetics
Let’s start with the basics: The birds and the bees and how we pass on our genes.

Our bodies are made up of billions of cells. We have skin cells, heart cells, brain cells, etc. These cells all have special functions in our bodies, but one thing they have in common is that each cell has a set of 46 chromosomes, or 23 pairs. The first 22 pairs of chromosomes are the same in men and women and the 23rd pair is different. Women have two ‘X’ chromosomes for their 23rd pair, and men have one ‘X’ chromosome and one ‘Y’ chromosome (see picture to the right of a set of male/female chromosomes, also called a karyotype).
Along the chromosomes are the individual instructions, or genes, that tell our bodies how to grow and function, including things such as hair color, eye color, and height. Since we have two copies of every chromosome, we also have two copies of every gene. All in all, it is estimated that we each have about 20,000 pairs of genes, one set from our mother and one set from our father. Males only have one X chromosome though, so they only have one copy of all of the genes on the X chromosome, and one copy of the genes on the Y chromosome.
Genes are specific instructions that contain our genetic code, or DNA (deoxyribonucleic acid). DNA is made up of four similar chemicals, called bases: adenine (A), thymine (T), cytosine (C ), and guanine (G). The specific order of these letters (bases) is what makes the instructions (genes) work properly. If one or more of these letters is changed, deleted, or duplicated, and this change causes the gene to not work as usual, this is called a mutation. Mutations in our genes may lead to genetic conditions in ourselves or other family members.
Many of the genetic conditions that are discussed in prenatal genetics are due to a baby having an extra (called a trisomy) or missing (called a monosomy) chromosome. An important thing to remember is that we have no control over how many chromosomes are packaged in our egg or sperm cells. If a baby has an extra or missing chromosome, there is nothing that was done to cause it and nothing that could have been done to prevent it.
Other types of genetic conditions involve changes in the instructions (genes) that are on the chromosomes. Some of the more common genetic conditions involving a single gene can be categorized into the three main ways that they can be passed down in a family: autosomal recessive, autosomal dominant and X-linked. These three terms are also referred to as ‘patterns of inheritance‘, and understanding the basics behind these terms will help to understand how genetic conditions may run in a family.
Because each gene does a specific job in the body, if someone inherits a non-working gene(s), what job that gene(s) does in the body generally determines what the genetic condition will be. For example, if someone inherits a non-working copy of a gene that is responsible for helping to build bone, then that person may have a genetic condition that causes differences with the bone development, while someone who inherits a non-working copy of a gene that helps to form structures in the ears may be hard of hearing.
Inheritance Patterns
Genetic, or hereditary, conditions can be passed down in a family in many different ways:
We have over 20,000 genes in our bodies, and each gene performs a very specific job. Some genes determine our hair color or our height, while other genes control how our bones and organs form and function.
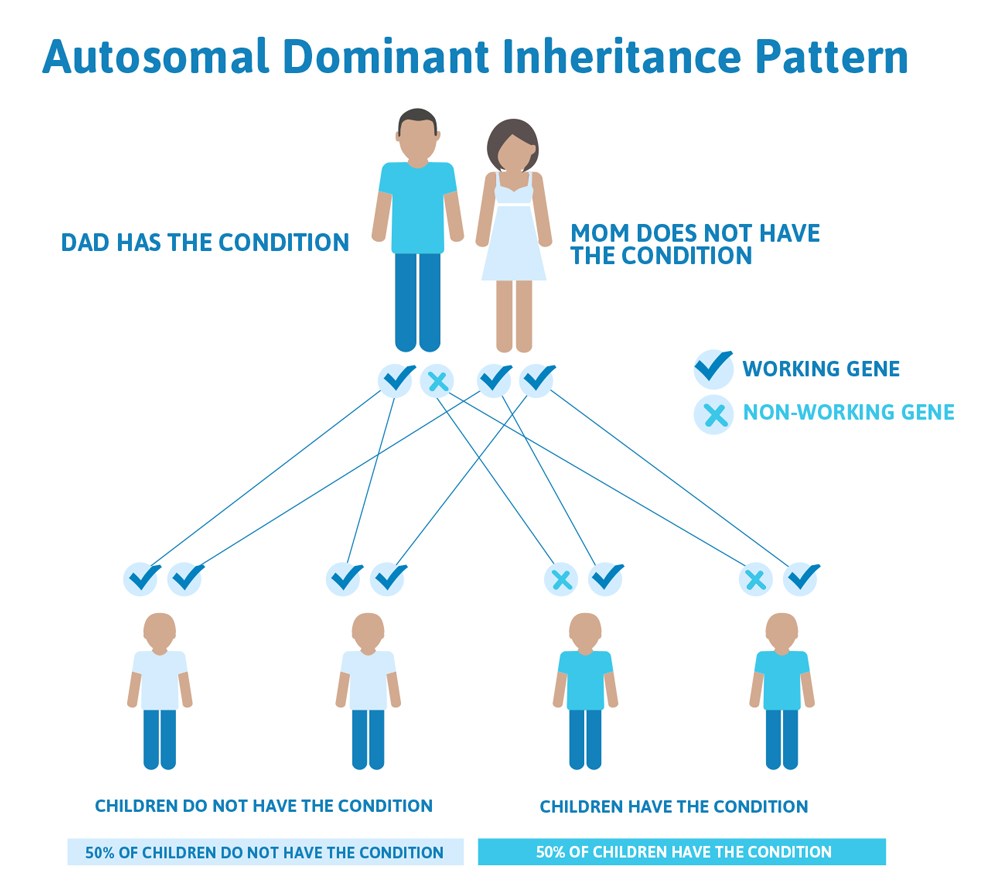
Typically, both copies from the pair are functional, but we all have genes that are not working how they should. In fact, scientists estimate that we each have approximately 20 non-working genes! This usually doesn’t cause a problem because you have two, so there’s a built-in backup system; if one doesn’t work the other one takes over. However, this is not true for all genes. There are many genetic conditions where if one gene of the pair is not working, the other one cannot make up for it and it can cause a genetic disease. This illustrates a common pattern of heredity called autosomal dominant inheritance.
Autosomal means the gene that is involved is found on one of the first 22 chromosomes (called the autosomes) and not on the X or Y chromosome (the sex chromosomes).
Dominant refers to the above explanation that we have two copies of each gene, one from mom and one from dad, and in order to have an autosomal dominant condition, a person only has to have one copy of the gene not working. They can inherit this copy from mom or dad, who may also have the condition.
So how does an autosomal dominant condition look in a family?
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. It is the same 50% chance for each pregnancy.
There are cases of autosomal dominant gene changes, called pathogenic variants, where no one in the family has it before and it appears to be a new thing in the family. This is called a de novo pathogenic variant. For the individual with the condition, the chance of their children inheriting it will be 50%. However, other family members are generally not likely to be at increased risk.
This diagram illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
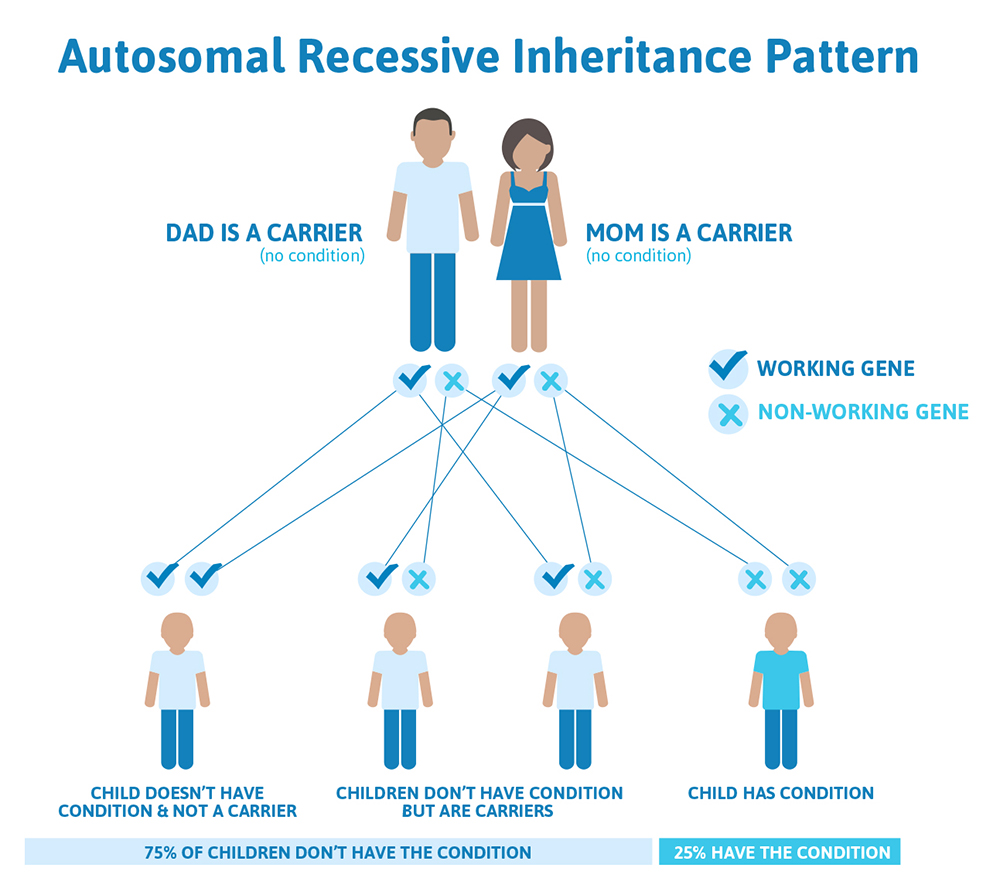
We have over 20,000 genes in our bodies, and each gene performs a very specific job. Some genes determine our hair color or our height, while other genes control how our bones and organs form and function. Typically, both copies from the pair are functional, but we all have genes that are not working as they should. In fact, scientists estimate that we each have approximately 20 non-working genes! This usually doesn’t cause a problem because you have two, so there’s a built-in backup system; if one gene doesn’t work then the other one takes over. However, if both copies of a gene that a person inherited do not work, this can lead to a genetic condition that is caused by autosomal recessive inheritance.
Autosomal means the gene that is involved is found on one of the first 22 chromosomes (called the autosomes) and not on the X or Y chromosome (the sex chromosomes).
In order to have a recessive condition a person has to have BOTH copies of a gene that are not working. The copy they inherit from mom is not working AND the copy they inherit from dad is not working, resulting in zero functioning copies of that gene. With recessive conditions, if you have one copy of the non-working gene (called a carrier), you do not have the condition and typically do not have any related symptoms.
How does an autosomal recessive condition look in a family?
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Most autosomal recessive conditions are individually rare, so the chance that both people are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same genetic condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, which would lead to the baby having the genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
That also means there’s a 50% chance the baby would be a carrier just like it’s parents, and as discussed above carriers generally do not have any symptoms. There’s also a 25% chance that the baby would inherit a working copy of the gene from both parents and not be a carrier.
We have over 20,000 genes in our bodies, and each gene performs a very specific job. Some genes determine our hair color or our height, while other genes control how our bones and organs form and function.
Most of our genes come in pairs; we get one from our mom and one from our dad. Typically, both copies from the pair are functional, but we all have genes that are not working how they should. In fact, scientists estimate that we each have approximately 20 non-working genes! This usually doesn’t cause a problem because there are two, so there’s a built-in backup system; if one gene doesn’t work the other one takes over. However, this is not true for all genes. There are many conditions where if one gene of the pair is not working, the other one cannot make up for it and it can cause a genetic disease.
While we usually have two copies of each chromosome, and therefore two copies of each gene, there is one considerable exception: our sex chromosomes.
Our 23rd pair of chromosomes are called the sex chromosomes. Women generally have two ‘X’ chromosomes (and therefore, 2 copies of all the genes on the X chromosome) and men have one ‘X’ chromosome and one ‘Y’ chromosome (and therefore, only one copy of the genes on the ‘X’ chromosome). This is helpful to know as we talk about X-linked inheritance.
X-linked simply means that the gene involved in these genetic conditions is found somewhere on the X chromosome. X-linked conditions are passed down in two ways: X-linked dominant or X-linked recessive.
X-linked dominant means that one broken copy of a gene on the X chromosome is enough to cause the genetic condition. Men only have one copy of their X chromosome, and therefore only one copy of all the genes that are on the X chromosome. If they inherit an X-linked dominant condition, they will be affected. There are several X-linked dominant genetic conditions that can be lethal to male pregnancies. For women with X-linked dominant conditions, they have one broken copy of the gene and one working copy of the gene. However, since only one broken copy of the gene is enough to cause the genetic condition, women who inherit a non-working gene for X-linked dominant conditions often also show symptoms (their backup working copy of the gene on the other X chromosome is not enough to make up for the broken copy).
X-linked recessive means different things for men and women. Women have two X chromosomes, so if they inherit a X-linked recessive condition they usually have a working back-up copy of the gene on their other X chromosome (and are called carriers). Because of this, often times women who are carriers for X-linked recessive conditions do not show any symptoms, although there are exceptions to this. Women who are carriers for X-linked recessive conditions and show symptoms usually have much more mild symptoms than are found in men. Women generally would need to have inherited non-working genes from both parents to be affected with an X-linked recessive condition.
Men, however, only have the one X chromosome. If a gene on their X chromosome is not working, they do not have another X chromosome to provide a back-up (instead of a second X chromosome, they have a Y chromosome). Therefore, if an X-linked recessive gene is passed down to a male, they will be affected with that genetic condition.
How does an X-linked condition look in a family?
X-linked conditions can be confusing in terms of how they are passed on and who gets the condition within the family. This is due in part because men have one X chromosome and women have two X chromosomes, but also because the chances for children to inherit X-linked conditions depends on which parent has the non-working copy of the X-linked gene: mom, dad, or it could be both!
Sometimes X-linked genetic conditions do not fit perfectly into either X-linked dominant or X-linked recessive. There are many other factors that can influence how these genetic conditions show up in a family, but these are some good general rules. Here are some scenarios that can help to further understand X-linked inheritance:
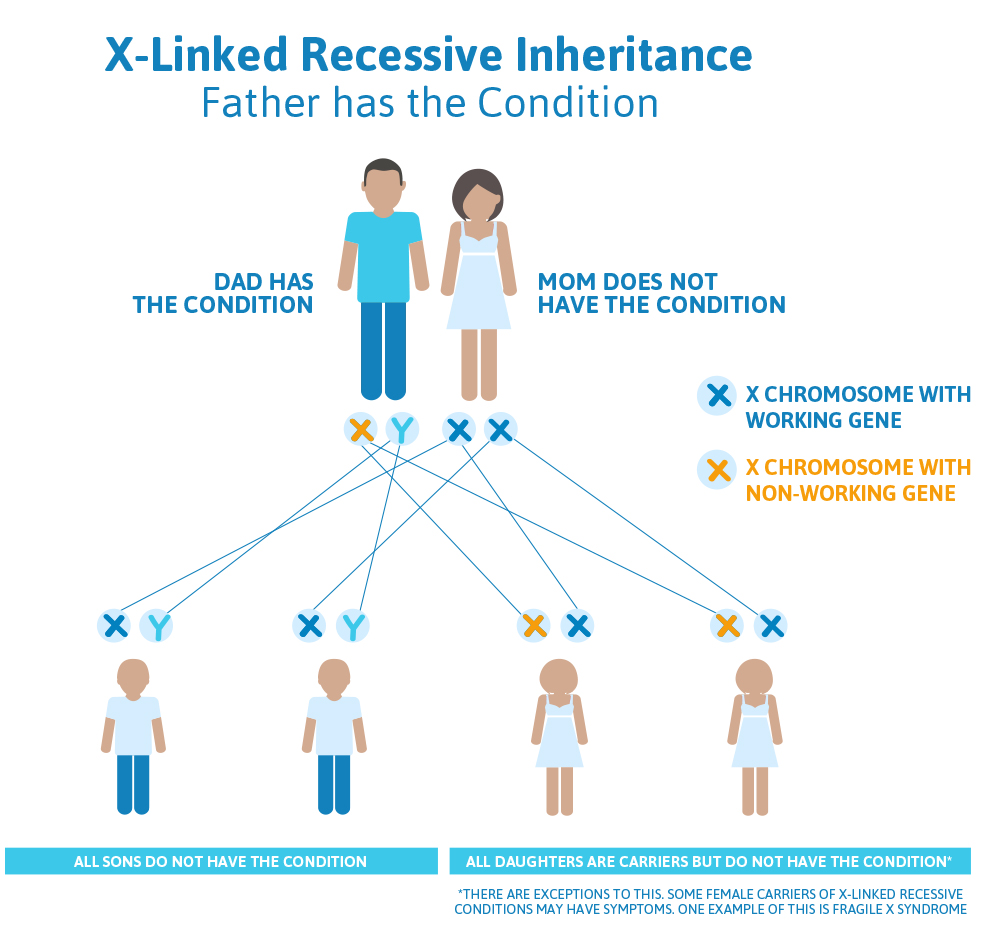
Scenario 1 (X-linked recessive): Dad has condition, Mom doesn’t
A man and his wife come in to see their doctor. The man tells his doctor that he was previously found to have an X-linked recessive genetic condition called Superman syndrome, but his wife was found not to be a carrier for it, and they have questions about what this means for their plans to have kids.
What we know:
- Because the man has Superman syndrome, and we know it is X-linked recessive, we know that his one and only copy of the Superman gene (which is on the X chromosome) is not working.
- Because the woman is not a carrier, both of her Superman genes (remember: she has two because she has two X chromosomes) are working.
What does this mean for their children?
- The chances to have a child who also has Superman syndrome depends on whether that child is a boy or a girl.
- The man will always pass down his Y chromosome to his male children, so none of his sons will be affected with Superman syndrome.
- Men have one X chromosome and one Y chromosome, while women have two X chromosomes.
- Women always pass down an X chromosome; if the man passes down his X chromosome, the baby will be female; if the man passes down his Y chromosome, the baby will be male.
- The man will always pass an X chromosome with a non-working Superman gene down to his daughters, because that is the only X chromosome he has; so, all of his daughters will be carriers for Superman syndrome.
- Carriers for X-linked recessive conditions have one gene on their X chromosome that is working and the same gene on the other X chromosome that is not working.
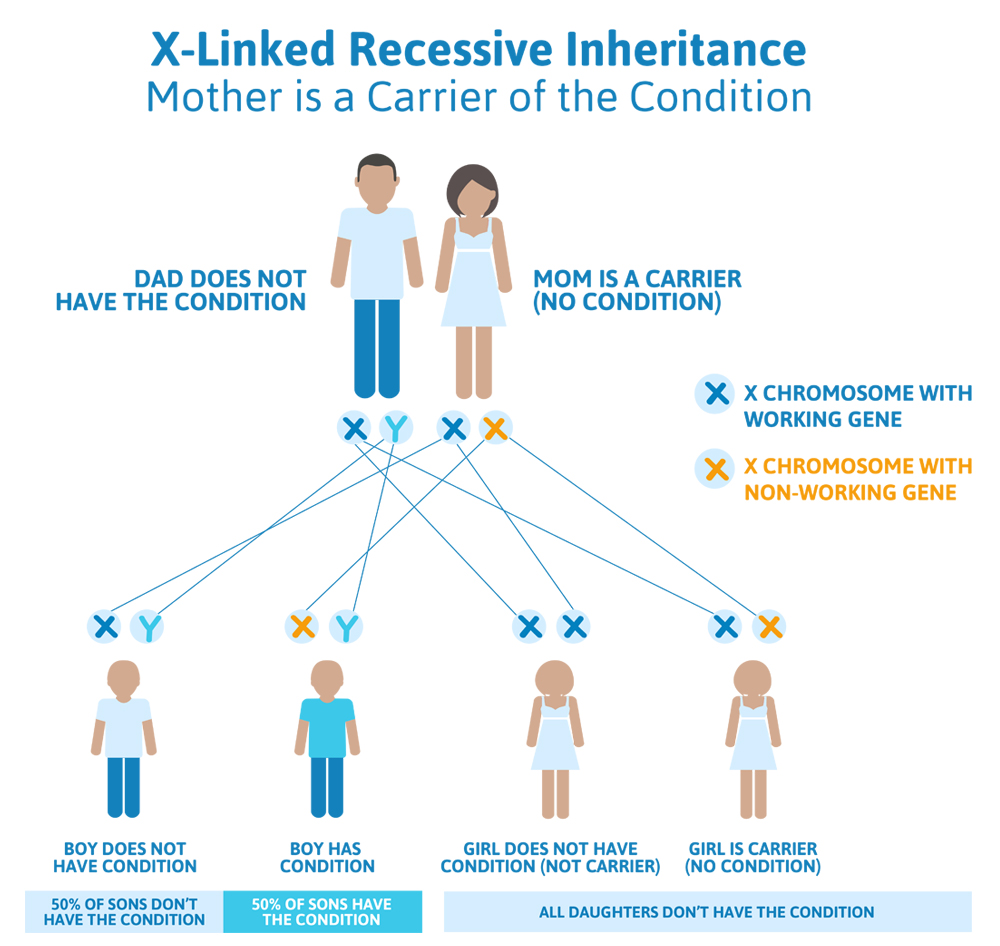
Scenario 2 (X-linked recessive): Mom is a carrier, Dad doesn’t have the condition
The daughter of our patient above comes into her doctor and tells him that her father had Superman syndrome, and that she was tested and confirmed that she is a carrier. Her partner was tested and does not have Superman syndrome, but they have questions about what this means for their plans to have children.
What we know:
- Because the woman is a carrier for Superman syndrome, we know she has one broken copy of the Superman gene (on one of her X chromosomes) and a working copy of the other Superman gene (on her other X chromosome).
- Because the man does not have Superman syndrome, we know that his one and only Superman gene is working.
What does this mean for their children?
- The chances to have a child who also has Superman syndrome depends on whether that child is a boy or a girl.
- The woman has a 50% chance to pass down the X chromosome that has the working Superman gene, and a 50% chance to pass down the X chromosome that has the non-working Superman gene to each child.
- The man has a 50% chance to pass down his X chromosome (that has a working copy of the Superman gene and produces a daughter), and a 50% chance to pass down his Y chromosome (that doesn’t have a Superman gene and produces a son).
- There are four different possibilities; each of these possibilities has a 25% (or 1 in 4) chance of happening:
- The woman passes the working copy of the Superman gene down, and the man passes down the X chromosome, giving them a girl that does not have Superman syndrome and is not a carrier for Superman syndrome.
- The woman passes the working copy of the Superman gene down, and the man passes down the Y chromosome, giving them a boy that does not have Superman syndrome.
- The woman passes the non-working copy of the Superman gene down and the man passes down the X chromosome, giving them a girl that does not have Superman syndrome but is a carrier for Superman syndrome.
- The woman passes the non-working copy of the Superman gene down and the man passes down the Y chromosome, giving them a boy that has Superman syndrome.
Scenario 3 (X-linked dominant): Dad has the condition, Mom does not
A friend of our couple above comes into his doctor because he found out that he has Batman disease, which is X-linked dominant. His wife has been tested and does not have Batman disease. They are concerned about the chances to pass on Batman disease to their future children.
What we know:
- Because the man has Batman disease, and we know it is X-linked dominant, we know that his one and only copy of the Batman gene (which is on the X chromosome) is not working.
- Because the woman does not have Batman disease, we know that both of her Batman genes (one on each of her X chromosomes) are working.
What does this mean for their children?
- Because both of the woman’s Batman genes work, there is a 100% chance that she will pass down a copy of a working Batman gene to any children they have
- The man has a 50% chance to pass down his Y chromosome to a child, which would give them a boy that does not have Batman disease.
- The man has a 50% chance to pass down his X chromosome (which carries his broken Batman gene), which would give them a girl who does have Batman disease.
- Given the inheritance pattern of this condition, this couple would have no chance to have a son with Batman disease. However, 100% of their daughters will have Batman disease..
Scenario 4 (X-linked dominant): Mom has the condition, Dad does not
The sister of the dad in the couple above comes into her doctor because she found out that she also has Batman disease, which is X-linked dominant. Her husband does not have Batman disease. They are concerned about the chances to pass on Batman disease to their future children.
What we know:
- Because the woman has Batman disease, and we know it is X-linked dominant, we know that she has one X chromosome with a working Batman gene, while her other X chromosome has a non-working Batman gene.
- Because the man does not have Batman disease, we know that he has a working copy of the Batman gene on his only X chromosome. His Y chromosome does not have the Batman gene on it.
What does this mean for their children?
- Because one of the woman’s Batman genes work and one doesn’t, there is a 50% chance that she will pass down a copy of a working Batman gene to any children they have, and a 50% chance to pass down a non-working copy of the Batman gene.
- Because the man does not have Batman disease, we know that if he has a daughter, he will pass down his X chromosome that has a working Batman gene.
- Because the Batman gene is X-linked, if the man has a son, he will pass down his Y chromosome, which does not have the Batman gene on it.
Thus, each pregnancy for this couple will have a 50% chance to have Batman disease.
Our cells have many different parts, and each one performs a very important function. The mitochondria is the energy, or power source, for the cell, and is what keeps the cells going and the body functioning how it should.
The mitochondria have their own set of DNA. The DNA in the mitochondria is made up of separate genes, or instructions, for how the mitochondria makes energy and keeps the cells going.
The DNA in our cell is what tells everything in our body how to work, from how tall we will be to the development of all of our organs and body systems. We inherit 50% of our cellular DNA from our mother, and 50% of our cellular DNA from our father. Our mitochondrial DNA is passed down only from our mothers. Therefore, a child can only inherit a mitochondrial condition from their mother.
Mitochondrial conditions are very complex. A specialist, such as your doctor or a genetic counselor, can provide more information and assessment if you are concerned about a mitochondrial condition running in the family.
Many of the most common health conditions, such as heart disease and stroke, are considered to be multifactorial. This means that there are many things that determine whether or not someone will develop that specific medical condition.
Diabetes, for example, is likely caused by a mixture of genetics, lifestyle choices, and environment. However, how much genetics affects someone’s risk may vary family to family. For example, if Jay has three relatives who have diabetes, his risk may higher to develop diabetes than Anne, who has no family history of diabetes.
With multifactorial conditions, heredity may offer some information as to someone’s risk, but it is complex, and our understanding of these health issues is constantly evolving. Some common multifactorial conditions are:
- heart disease
- high blood pressure
- Alzheimer’s disease
- arthritis
- diabetes
- obesity
- cancer
- high blood pressure
- high cholesterol
- thyroid disorders
- some birth defects, such as cleft lip and/or palate, spina bifida, and heart defects
Some families may have a more significant genetic cause to their health issues, particularly with certain types of heart disease or cancer. A genetic counselor can review your personal and family history and can provide more insight into what your specific risk might be.
Family History
When it comes to genetics, it’s not all about you; your entire family’s health history is important. There are many health conditions where someone’s risk to also develop that condition is determined by their family history. Because of this, it is important to try to gather as much information about your family health history as possible.
In general, it’s good to have information about your kids, grandkids, brothers and sisters, nieces and nephews, parents, aunts and uncles, cousins, and grandparents. Any information for relatives beyond that may also be helpful to have, particularly if they have any known health issues. It may be helpful to write this information down to have record of it and to share with other family members.
Any health condition is good to note, but pay extra attention to anyone with:
- A genetic condition (like cystic fibrosis, Tay Sachs, or sickle cell anemia)
- A chromosome condition (like Down syndrome, trisomy 18, or Turner syndrome)
- Intellectual disability/delay
- Birth defects (cleft lip and/or cleft palate, club feet, heart defect, etc.)
- Loss of sight and/or hearing from birth
- Babies that passed away at birth or during infancy
- Unexplained childhood illnesses or deaths
- Multiple miscarriages (three or more)
- Infertility, or unable to have children
- Cancer
- Important to note the type of cancer (breast, colon, uterine, etc), and age of diagnosis
- Heart attack at an early age (before age 55 in men and before 65 in women)
- Ethnic backgrounds
- Some genetic conditions are more common in certain ethnic backgrounds, so it is important to try to find out what country your family comes from originally
- Other health conditions that seem to run in your family that you are curious or concerned about
What is a pedigree?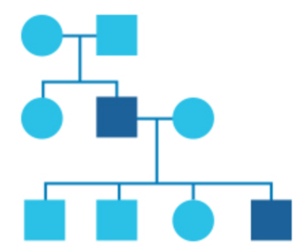
In genetics, family history information is recorded in a diagram called a pedigree.
In this diagram, circles represent women and squares represent men. Drawing out your family in this way can help identify patterns and provide a clearer picture of any health issues running in your family.
There are many reasons that someone may not have a lot of information about their family history: adoption, family dynamics, people move to different parts of the country and fall out of touch, etc. To get a full picture of someone’s risk for various health problems, it is important to have as much information as possible. When practical, it may be helpful to reach out to family members, or in cases of adoption, contact the adopting agency to see if there is any health information on file.
Having these conversations with relatives can be challenging at first. It may be helpful to begin by talking with a family member that you feel comfortable with to start gathering information. Talking to other relatives can help to fill in any gaps in information. It is important to feel as comfortable as possible having these conversations. One way to engage family members may be to point out that once all of this information is gathered in one place, it will be beneficial to everyone in the long run.
The March of Dimes has a general family history form that may be helpful to use to start gathering all of this information.
- Keep a copy in a safe place
- Share it with your doctor.
If you are concerned about anything that you’ve learned in your family history, you can reach out to a genetic counselor, search here.
Genetic Testing
Genetic testing is a rapidly changing area of medicine, and whether or not to undergo genetic testing may not be an easy or straightforward decision. Even if you have already undergone genetic testing, the results may often be confusing to interpret. A genetic counselor is a medical professional that can help answer any questions you have, make sure you have all of the information you need to make a decision that is best for you and your family, and help make sense of genetic testing results. Select a topic below to learn more about genetic testing.
A chromosomal microarray (CMA) is a test that can be done to look for specific kinds of chromosome changes called microdeletions and microduplications.
Much like a traditional chromosome analysis, CMA generally looks at all 23 pairs of chromosomes. While the chromosome analysis is looking for large changes (extra or missing pieces of chromosomes, or extra or missing whole chromosomes), the CMA testing can look for much smaller missing or extra pieces of chromosome material.
It can be difficult to understand how each test is similar and different, so let’s look at it a different way. Imagine a bookshelf full of books. A traditional chromosome analysis would be like looking at the shelf, and figuring out if there are any extra or missing books, or if the books or in the wrong order. CMA testing would be like taking a book off of the shelf and looking through it to see if there are any missing or extra pages. Traditional chromosome analysis can find large differences, while CMA can take a more in-depth look.
CMA testing can be done for a number of reasons, including for children and adults who may have an underlying genetic condition, but it’s not clear what that genetic condition is. Each genetic condition is related to specific genes. If we have an idea of what genetic condition someone might have, then we can offer them testing that is more targeted to what we think is most likely causing it. If someone has health issues that could have a genetic cause but are not specific (some examples may include developmental and intellectual delays, or autism spectrum disorder), CMA testing may be used to take a wider look at a patient’s chromosomes to try to determine if an underlying genetic cause can be found.
CMA testing can also be done during pregnancy; click here to read more.
Because test results from CMA testing may not necessarily be straightforward, it is important to talk with healthcare provider who is familiar with CMA testing who can explain more about the risks, benefits, and limitations of the testing.
A chromosome translocation, also called a chromosome rearrangement, is when pieces from two separate chromosomes break off and switch spots. We know from Genetics 101 that each cell of our body contains chromosomes that act as an instruction manual, telling our body how to grow, develop, and function. We usually have 23 pairs of chromosomes (one set coming from mom and the other set from dad), which gives us a total of 46 chromosomes. The body is very particular in the amount of this information that we have, and too many or too few of these instructions can change how things work in the body.
Chromosome translocations can run in families (inherited from a parent), or a person can be the first one in their family found to have it (new to that person, called de novo). If someone is the first one in their family to have it, that means that the chromosome translocation likely happened early in their development. We do not yet know what causes chromosome translocations to happen.
Most of the time when a person is found to have a chromosome translocation, it is classified as a balanced translocation. A balanced translocation means that all of the instructions are still in that person’s cells, they’re just in a different order. Most often, balanced translocations do not cause any significant health issues. However, if someone has a balanced translocation, they are at an increased risk to pass on too much or too little chromosome material to any children they may have.
For example, let’s say someone has the balanced translocation between the blue chromosome (B) and the green chromosome (G), shown below:
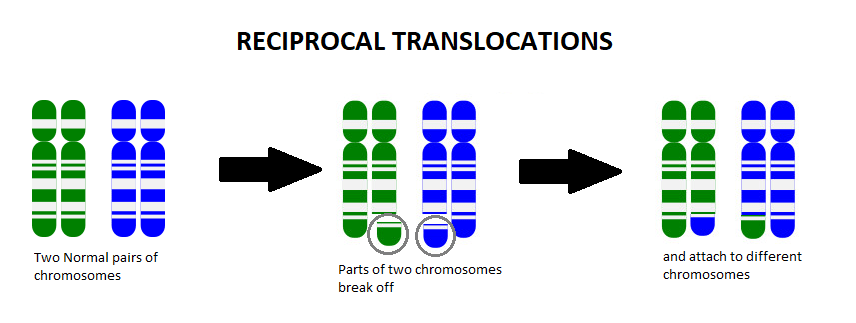
Someone who has this balanced translocation has one whole G chromosome, one whole B chromosome, one mostly G chromosome with a small amount of B on it (G+B), and one mostly B chromosome with a small amount of G on it (B+G).
Because parents pass on one copy of each of their chromosomes to each child they have, each pregnancy has a 50% chance to inherit the whole G chromosome, and a 50% chance to inherit the G+B chromosome. Each pregnancy would also have a 50% chance to inherit the whole B chromosome, and a 50% chance to inherit the B+G chromosome.
![]()
If a child inherits a whole G chromosome and a whole B chromosome from one parent, but the G+B chromosome and a whole B chromosome from the other parent, they will have too much B chromosome material and not enough G chromosome material.
The specific extra B instructions and missing G instructions will determine what sort of health problems can result. For example, if someone is missing part of a chromosome that had instructions for how to form connections in the brain, they may have a higher risk for health problems related to their brain function, such as seizures or learning delays.
We have over 20,000 genes in our body, and each gene has a very specific job. To do that job, our genes make substances called proteins that make everything in the body work, grow, and function properly. A gene is made up of a long string of many letters (A, C, G, and T) that act as an instruction manual for how to make a specific protein.
Much like spelling errors (sequence variants) can affect how a gene makes a protein, so can extra and missing letters, which are called duplications and deletions. Adding or removing letters from an instruction manual affects how the instructions read, and can also cause the gene to make either an abnormal protein or no protein at all, which is what can cause genetic conditions.
A similar example is self-assemble furniture. Let’s say you buy a kit at the store to build a jungle gym for your kids in the backyard. You lay all of the pieces out, start going through the instructions and putting things together. When you get done, the jungle gym doesn’t look like it’s supposed to and you have a bunch of extra pieces left over. You go back through the instructions and realize that there is a page that is missing. Without that missing page, you don’t have proper instructions on how to put together your jungle gym. The instructions for how a gene builds a protein is very similar.
Some missing or extra pieces (called variants) do not cause a problem. If you’re looking at your jungle gym instructions and it turns out you have a duplicate of one of the pages. It’s not necessary, but it’s also not hurting anything. Whether the missing or extra piece in a gene causes a problem depends on the gene, and where in the gene the extra or missing piece is at.
Variants in genes can be broken down into five different categories:
- Pathogenic variants are known to make the gene not work correctly, and can cause a genetic condition.
- Likely pathogenic variants probably make the gene not work correctly, but more information is needed to say for sure.
- A variant of uncertain significance means there is not enough information to say whether the variant is harmful or not, and more research on that variant is needed.
- Likely benign variants probably don’t impact how the gene works, and thus likely does not cause a genetic condition.
- Benign variants are known to not impact how the gene works, and do not cause a genetic condition.
Genetic testing for someone who may be at risk for an inherited disease is always easier if we know the specific genetic cause. Often times, the best way to find the genetic cause is to start by testing someone in the family who is known or strongly suspected to have the disease. If their testing is positive, then we can say that we have found the familial pathogenic (harmful) variant. We can use this as a marker to test other members of the family to see who is also at risk.
An example of this may be a woman who comes in to see a genetic counselor because she has a family history of breast cancer. Her mother had breast cancer at 42, which is considered early-onset, and had recently gotten genetic testing and found that she has a pathogenic variant in the BRCA1 gene, which causes hereditary breast and ovarian cancer syndrome (HBOC). Individuals with HBOC have a higher chance to develop certain types of cancer, particularly breast and ovarian cancers in women. This means that our patient has a 50% chance to also carry that same pathogenic gene variant. Because we were able to find what the specific cause is for the increased cancer risk in her family:
- if our patient tests negative for the familial pathogenic variant, we will be able to tell her that she should have the same risk as the average woman for breast cancer and ovarian, regardless of her family history
- if she tests positive for that pathogenic gene variant, then we know she is at an increased risk for breast and ovarian cancer, and we can discuss different pathways to help keep her healthy
Who the best person to start genetic testing within the family to try to identify a familial pathogenic variant is complex, and depends on what the condition is that you are testing for and the inheritance pattern, as well as your family history. Genetic testing also may not be indicated or helpful, depending on the medical concern.
Testing for a familial pathogenic variant can also be done prenatally, or during pregnancy. If one or both parents are found to be carriers for a genetic condition, a procedure such as a CVS or amniocentesis can be used to get fetal cells that can then be tested. These procedures come with some risk of miscarriage, so it is important to talk with your doctor or genetic counselor to make sure you have all of the information you need to make the decision that is best for you and your family.
A healthcare professional with a background in genetics, such as a genetic counselor, can help you assess all of this information and provide support.
Mosaicism is a term that describes when two different types of cells can be found in one person. We know from Genetics 101 that each cell of our body contains chromosomes that act as an instruction manual telling our body how to grow, develop, and function. We usually have 23 pairs of chromosomes, one set coming from mom and the other set from dad, which gives us a total of 46 chromosomes. The body is very particular in the amount of this information that we have, and too many or too few of these instructions can change how things work in the body.
In the simplest form of mosaicism, an individual may have 46 chromosomes in some cells of their body and 45 chromosomes (missing a chromosome) in some cells of their body. For example, most females have two X chromosomes, while most males have one X and one Y chromosome. However, there is a genetic condition called Turner syndrome in which females only have one X chromosome (they do not have a second X chromosome). Some females who have Turner syndrome actually have mosaic Turner syndrome, meaning that some cells in their body have two X chromosomes (not Turner syndrome), while other cells in their body only have one X chromosome (Turner syndrome). Mosaicism can happen in the case of a whole chromosome as noted above or can happen for a single gene, or single instruction.
People who have mosaic genetic conditions, such as Turner syndrome, do not generally have as many of the health concerns as someone who has the genetic condition in every single cell of their body. It can also be difficult to determine if someone has mosaicism, and if they do, which cells in the body may be affected. Even if it is determined that someone is mosaic, there is not an easy way to determine what percentage of their cells are ‘normal’ and what percentage of their cells are ‘abnormal’, and where in the body each type of cell is.
Next generation sequencing (aka panel testing) is a relatively newer technology that allows the lab to look at many different genes in a faster and more cost-effective way than ever before. This type of testing can be particularly helpful in situations where there are many potential explanations for what we see in a personal or family history.
Before this testing existed, doctors would go through the list of possible genetic conditions that explain the medical and family history one by one starting with the most likely. Test results for each gene would take anywhere from two to 12 weeks to come back, and the cost for testing per gene was anywhere from $500 to $5000!
Next generation panel testing can allow the lab to look at several genes at one time, and results are generally back within three to 12 weeks, depending on the type of testing and the lab. The price of panel testing can be up to several thousand dollars, but is on the decline for most types of tests. Given the price and time burden of testing each gene individually, in many cases it may be more time and cost-effective to consider a panel test.
That sounds amazing! Why wouldn’t I get panel testing?
While panel testing has shown to have many benefits, there are also some potential drawbacks to consider:
Uncertain Guidelines
Some labs are offering testing for genes, particularly some that are linked to an increased risk for cancer, that have no official guidelines. An example of this would be:
Ms. Abernathy was seen by her genetic counselor because she has a strong family history of breast cancer. The testing came back that she has a harmful change, or pathogenic variant, in a gene that some research studies show is likely related to an increased risk for hereditary breast cancer. However, not enough data has been gathered to show how high the risk is, or what medical management recommendations should be for women who carry a pathogenic variant in this gene. This can create a difficult situation because we have some information, but not enough to be able to tell someone definitively what to do next.
Variants of Uncertain Significance
Another potential concern with panel testing regards variants of uncertain significance (VUS). Visit our results page to learn more about VUS findings.
Unexpected Findings
Sometimes genetic testing can be ordered to look for an answer to a potential health concern, but can provide unexpected, although ultimately helpful, information.
Let’s say Ms. Abernathy from above was seen by her genetic counselor for a family history of breast cancer, and her genetic counselor ordered a panel test that looked to see if she has an increased risk for various different types of cancer. Her testing came back negative for all of the genes related to hereditary breast cancer, but they did find a pathogenic variant in a gene related to an increased risk for colon cancer.
So, while the test was not able to provide an explanation for her family history of breast cancer, it does give her information that she is at an increased hereditary risk for colon cancer. Ms. Abernathy and her doctor can use this information to begin colon cancer screening at an earlier age and repeat them more frequently than she would otherwise get them in order to keep her healthy.
Another concern with genetic testing is that it may reveal non-paternity (someone’s father is someone different than who they thought), which is thought to occur in 2-10% of all people. While thought to be relatively rare, certain types of genetic testing may bring this to light, potentially causing additional anxiety and stress.
We have over 20,000 genes in our body, and each gene has a very specific job. To do that job, our genes make substances called proteins that make everything in the body work, grow, and function properly. A gene is made up of a long string of many letters (A, C, G, and T) that act as an instruction manual for how to make a specific protein. These letters are called nucleotides.
In some parts of the instruction manual there are short strings of letters that repeat. These short strings of letters may include three (tri-), four (tetra-), five (penta-), six (hexa-), or more letters that repeat many times over. The number of repeats can vary between people even within the same family, but most people have a similar number of repeats. A repeat expansion is a genetic change where the number of repeats someone has increases in number. This can disrupt the instruction manual causing the gene to make either an abnormal protein, too much or too little protein, or no protein at all, which is what can cause some genetic conditions.
For example, a gene’s letter sequence may have a three letter repeat (also called a trinucleotide repeat) that spells CAT over and over again. When someone has a typical amount of repeats, the gene produces a normal protein.
CAT CAT CAT
= normal protein
Sometimes people can have extra repeats, but they don’t affect how the instructions read, and thus don’t create a problem.
CAT CAT CAT CAT CAT CAT
= normal protein
When someone has too many repeats, that can change how the body reads that gene’s instructions. This change impacts the protein that is produced, which is what can in turn cause some genetic conditions.
CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT
= abnormal, too much/little, or no protein
Genetic conditions caused by this type of change are called repeat expansion disorders. For these types of conditions, we typically know how many repeats someone would need to have in order for that individual to be affected. For some (not all) of these disorders, we also know individuals can show different signs or symptoms, or be diagnosed at different ages based on the number of repeats they have.
Compared to other types of genetic changes that often are the same between family members, repeat expansions can differ between family members. When there is a repeat expansion disorder in a family, family members may be affected differently based on the number of repeats they have. This is caused by what is called ‘instability’ of the repeat. It’s also possible to see future generations be more severely affected than prior generations due to the repeat expansion growing as it’s passed down in the family (called anticipation). For example, there may be a mother and daughter who are affected differently:
Mother =
CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT
= mild symptoms
Daughter =
CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT CAT
= severe symptoms
The language that is used to describe the different categories of repeats outside of what is considered normal or typical varies depending on what genetic condition you’re talking about. Possible descriptions may include:
- Normal/typical: repeat size is within the typical range; the gene is not impacted and works properly. These individuals are not affected.
- Mutable normal or intermediate: repeat size is on the higher side of the typical range; the gene is not impacted and works properly, but there is a higher risk the repeat could expand in future generations. These individuals are typically unaffected.
- Premutations or incomplete penetrance: repeat size is increased above what is typical, and the gene may be impacted. Some individuals may experience specific signs or symptoms, while others may not.
- Abnormal (also called full mutation or fully penetrant): repeat size is in a range where the gene is impacted and unable to work properly. All individuals with a full mutation will have signs or symptoms, but there can be variability in the severity (e.g. mild, classic, severe) and the age that someone is diagnosed.
We have over 20,000 genes in our body, and each gene has a very specific job. To do that job, our genes make substances called proteins that make everything in the body work, grow, and function properly. A gene is made up of a long string of many letters (A, C, G, and T) that act as an instruction manual for how to make a specific protein. If there are any spelling mistakes in that instruction manual (called a variant), it can cause the gene to make either an abnormal protein or no protein at all, which is what can cause genetic conditions.
Sequencing is the process where the lab reads through the letters of a gene and looks for any spelling errors that may affect how the gene works.
A sequence variant is when there is some sort of spelling error in the gene. There may be an ‘A’ where there is normally supposed to be a ‘C’. Some of these spelling errors happen in a part of the instructions that don’t affect how the instructions read and don’t create a problem (imagine an instruction that is supposed to say ‘THE CAT RAN’ but instead says ‘THE CAT RUN’. The grammar isn’t perfect, but the message is still pretty much the same). This is called a benign (non-harmful) variant.
Other spelling errors happen in a part of the instructions that change the whole meaning (an instruction that is supposed to say ‘THE CAT RAN’ but instead says ‘THE CAB RAN’. That one letter difference changed the entire meaning of the sentence). This is called a pathogenic (harmful) variant. Pathogenic variants in genes can affect how the instructions make a protein. If the protein cannot work how it is supposed to in the body, it can cause a genetic condition.
Variants in genes can be broken down into five different categories:
- Pathogenic variants are known to make the gene not work correctly, and can cause a genetic condition.
- Likely pathogenic variants probably make the gene not work correctly, but more information is needed to say for sure.
- A variant of uncertain significance means there is not enough information to say whether the variant is harmful or not, and more research on that variant is needed.
- Likely benign variants probably don’t impact how the gene works, and thus likely does not cause a genetic condition.
- Benign variants are known to not impact how the gene works, and do not cause a genetic condition.
A chromosome analysis, or karyotype, is the traditional testing that can been done on cells from chorionic villus sampling (CVS) and amniocentesis procedures, or from a blood sample. A karyotype involves the lab examining the cells and determining the number of chromosomes.
How the lab determines the number of chromosomes is a complicated process. Chromosomes all have bands on them, which kind of look like stripes. The pattern of the bands can help the lab determine which chromosome they’re looking at. For example, chromosome 21 has a very different banding pattern than chromosome 22.
These bands aren’t easily visible so the lab uses staining, much like you would stain a glass window, to show the pattern of bands on the chromosome. The lab can then use that pattern to figure out what number chromosome they are looking at. They then use a computer to put the chromosomes in order. Most people have two of each chromosome, so the lab would use the computer to move the chromosome number 1s together, and next the chromosome number 2s together, and so on.
Once all 23 sets of chromosomes are laid out we get a picture, which is the karyotype. This allows us to see if there are any extra (called a trisomy) or missing (called a monosomy) chromosomes. A karyotype can also identify if large pieces of chromosomes:
- Have broken off and switched spots (called chromosome translocations),
- Have broken off and turned upside down (called chromosome inversions),
- That are missing (called chromosome deletions), or
- That are extra (called chromosome duplications).
If there is a trisomy, monosomy, translocation, inversion, deletion, or duplication, it is important to determine what chromosome(s) are involved. Each chromosome has different information on it, so if there is extra or missing chromosome information, what is extra or missing can help us determine if it can cause health concerns.
Karyotype results are usually back within 2-3 weeks after the lab receives them. It is important to remember that a karyotype can be diagnostic for large chromosome differences, but cannot identify all genetic or hereditary conditions.
Whole genome sequencing (WGS) and whole exome sequencing (WES) are two of the most comprehensive genetic tests currently available. If you haven’t already, you may wish to review some of the basics in Genetics 101 and gene sequencing, as that information will be very helpful background to have first.
Background
The entire genetic code in humans is called the human genome. Our genome is made up of about 20,000 individual genes, or instructions. Our bodies read and process that information, and use the instructions to make proteins. These proteins carry out different functions or jobs within the body. More specifically, each gene is made up of two alternating types of information called “exons” and “introns”. Every gene contains multiple exons and multiple introns. When the body is processing a gene to make a protein, the introns are removed and it is mainly the exons that tell the body how to make the proteins. Most known genetic changes that cause human disease or health issues are found in exons. This is why the exons are considered the most important part of the genome, and why we used to refer to the introns as “junk DNA”. We now know that the introns are not junk, but we are still learning how information in the introns is used and what role they play in human development and function.
What is WGS and WES? And how are the tests different?
Through the use of technology called next generation sequencing, we are now able to look at large amounts of DNA at one time. With this technology, we can look at the letter sequence of all of the genomes exons. This is called the exome, and it makes up only 1-2% of the genome. This testing is called whole exome sequencing (WES). In comparison, whole genome sequencing (WGS) is when we look at all of the letter sequence of the entire genome (introns and exons).
WES is more commonly used in patient care for multiple reasons. At this time, we are able to better understand how variations in the letter sequence of an exon impacts our health. Also, WES generally costs less, and we are able to analyze the data more quickly.
WGS is sometimes used in patient care, but is more commonly used in research. In addition to looking at both introns and exons, WGS is able to better identify copy number variations (parts of the genome that are repeated – the number of these repeats may vary from person to person), and may be a more effective test for certain parts of the genome.
What type of results may be found?
All of us have variations in our DNA; it is part of what makes each one of us unique. As part of WES and WGS, hundreds to thousands of variants (changes) in the letter sequence may be found. The testing laboratory will analyze these variants and only provide information on a small amount of them because a majority of these variants do not have clinical relevance (they are part of what makes us unique and are not thought to cause health problems). These variants are classified as “benign” or “likely benign.”
For those who undergo WES or WGS to help identify a genetic cause for their complex medical history, the primary focus is generally on variations in genes that, after analysis and interpretation, are thought to be the underlying genetic cause for the that person’s medical concerns. It is important to know that not all variants that are reported will be in a gene that is associated with known or well-described health issues. There is a potential of a variant being reported in a gene that we don’t know much about yet (called candidate genes). The type of variants reported may be classified as “pathogenic” (known to cause or contribute to health concerns), “likely pathogenic” (strongly suspected to cause or contribute to health concerns), or “variant of uncertain significance” (it is unknown whether it causes or contributes to health concerns).
To help with the interpretation of these variants, the laboratory may use past medical history, family history, and previous test results. Also, family member’s samples may be requested (such as DNA from parents) to help understand if the variant is likely to be affecting someone’s health or not.
Please see our page on genetic testing results for more general information.
Secondary Findings
Because WES and WGS look at all of our genes, an individual may be found to have variant in a gene that causes a medical condition that is not related to why the testing was done. An individual may or may not already have symptoms of the condition. These type of results are called “secondary findings” or “incidental findings.”
The American College of Medical Genetics (ACMG) published recommendations that testing laboratories should report secondary findings in genes that are associated with medical conditions with clear and immediate medical significance. ACMG provided a list of 59 genes that should be reported when a “pathogenic” or “likely pathogenic” variant is found. This list of genes includes conditions such as hereditary cancer syndromes (i.e. high risk for cancer), conditions with significant heart disease risk, or others with known treatment or preventative recommendations.
Other secondary findings that may be reported by a laboratory include other known genetic disorders not associated with the individual’s medical concerns that are not on the ACMG list, carrier status for recessive disorders, and pharmacogenetic variants.
The reporting of secondary findings are optional. When you undergo informed consent for WES or WGS, you should be given the option to either opt-in or opt-out of these types of results.
Are there potential risks to doing this testing?
The primary potential risk associated with this testing is that the results may reveal genetic information about the patient or other family members that is unexpected (unrelated to the reason the testing was done). For example, a result may reveal that a patient or family member is affected with a specific genetic condition that may or may not have treatment or preventative recommendations.
Also, because we often request samples from other family members, we may inadvertently determine non-paternity (the person who was thought to be the father is not actually the biological father), or that an individual’s parents are related (called consanguinity).
Are there limitations to this testing?
Although both WES and WGS are becoming very important diagnostic tools in discovering the cause of rare disorders, there are limitations. These limitations can be somewhat different between WES and WGS. There are some parts of our genome that are difficult to sequence. This may be due to the specific letter sequence, or the location of the letter sequence in the gene. Some types of genetic changes cannot be found by these technologies.
What does it mean if my WES or WGS was “negative”?
A negative result does not rule out an underlying genetic cause for an individual’s health concerns. Variants could be found in the DNA that were not reported because based on our knowledge at this time, they were not suspected to be associated with any health issues. Over time, we will likely learn more about these variations. In the future, you can request reanalysis of the data to see if additional information can be provided.
Genetic Testing FAQs & Concerns
Genetic testing for cancer risk is a very personal decision, and is not right for everyone. Some people are concerned about the benefits versus potential risks, while others are more concerned with insurance or privacy issues. Select a topic below to learn more about these common concerns pertaining to genetic testing.
Healthcare costs are a very relevant part of decision-making with regard to whether or not to have any type of testing done.
Many insurance companies cover genetic testing when it is indicated, but each insurance company and plan is different. Some insurances may cover genetic testing but require a prior authorization (may also be called a prior authorization or precertification), which your genetic counselor can also help with. Each insurance company has their own policy on whether or not they will approve genetic testing and, if so, how much they will cover. These policies are constantly changing, particularly now as genetic testing becomes much more common and new tests and technologies are available. The best way to determine if you have coverage is to reach out to a genetic counselor or other knowledgeable provider to help you through this process.
If you do not have insurance coverage, there are options available that may help you cover the cost of genetic testing. As genetic testing technologies improve and testing becomes more integral to medical care, the costs of genetic testing have steadily declined. However, costs range widely (from $100 to thousands of dollars) depending on the laboratory and type of testing that is ordered. Many testing labs offer patient assistance programs that can help to cover the cost of genetic testing. Your genetic counselor may also know of other resources to cover or mitigate the cost (it never hurts to ask!).
The great majority of the states have had laws in place for many years to protect against genetic discrimination in health insurance and/or employment. In 2008, the Genetic Information Nondiscrimination Act (GINA) was signed, which is a federal law protecting against health insurers (group and individual health plans) using or requiring genetic test results to determine eligibility, coverage, or premiums. Notably, this law protects against the adverse use of genetic test results in the pre-symptomatic setting (e.g. having a high risk for cancer), but does not extend to protection in cases where the individuals already have symptoms of the condition (e.g. having a cancer diagnosis). This law does not apply to Tricare, the Federal Employee Health Benefits Plan, or the Indian Health Service. Additionally, there are not currently laws in place to protect against life insurance, long-term care, or disability insurance from requesting or using genetic information.
GINA also protects against employment discrimination based on genetic information, such as hiring, firing, pay, or other employment conditions.
The Affordable Care Act of 2010 includes a provision that provides insurance coverage with no out-of-pocket costs to patients for certain preventative healthcare services that are approved by the United States Preventive Services Task Force (USPSTF). In 2013, the USPSTF came out with updated guidelines on genetic counseling and testing of the BRCA1 and BRCA2 genes that recommends genetic counseling and testing for unaffected women with a family history suggestive of a possible pathogenic (harmful) variant in these genes. If you meet the criteria stated in the USPSTF guidelines, your insurance is likely to cover your genetic counseling and testing with no out-of-pocket cost to you.
The USPSTF guidelines do not address a few important areas:
- As these guidelines are meant for preventative medicine, and recommendations are meant to focus BRCA counseling and screening in the general population to those to be considered at higher risk, the focus of these guidelines center on testing unaffected individuals in the family when, in many cases, genetic testing is most informative when performed on an individual in the family who has been diagnosed with cancer (see the ‘Information for Family Members’ section of the Why Consider Genetic Testing? page for more information). Furthermore, the USPSTF maintains that their recommendations do not hold to women with a history of breast or ovarian cancer, even if it is a past history and they currently have no evidence of disease and are ‘unaffected’.
- The patient population covered by the guidelines also does not include women who have a known BRCA1 or BRCA2 gene pathogenic variant in their family.
Click below for more resources related to the Affordable Care Act:
- USPSTF recommendations
- Affordable Care Act genetic counseling and testing recommendations
- FORCE (Facing Our Risk of Cancer Empowered) is a nonprofit organization who put out a response to the USPSTF recommendations and some of the limitations.
We discussed in ‘Why Consider Genetic Testing’ several ways in which genetic testing may change recommendations for patients and their families. However, these reasons are not universal and may not apply to your situation. For example, your treatment and/or screening plan may not change based on genetic testing. Or you may not have children or other relatives who would benefit from your genetic testing results. For some people, the knowledge alone is of interest while for others it offers no clear benefits or is simply not desired. The decision of whether or not to have genetic testing is very personal, and should only be made after gathering all of the information that you would like to have.
The information that comes from cancer genetic testing can be stressful and anxiety-inducing for some. You are encouraged to openly discuss your concerns with your genetic counselor/qualified provider, but also with your family to define your current needs, clarify any misconceptions, and decide whether genetic testing is or is not going to be beneficial for you at this time. If you decline genetic testing, you can always revisit the option at a later time if you choose. In the absence of genetic testing, it remains important to follow the treatment/screening recommendations based on your personal and family history.
In some cases, the likelihood of receiving an uncertain result is greater than receiving any definitive answers on genetic testing (this is particularly likely on the larger gene panel tests that include multiple “newer” genes). Uncertain results can cause confusion and distress, and until they can be interpreted with a greater degree of certainty may not change any recommendations for you or your family. Faced with this possible outcome, some people may choose to defer testing until more information becomes available.
Whether or not to pursue any genetic testing is a very personal decision. In some cases, moving forward with genetic testing may help to provide an answer to a health question that is running in someone’s family, or may help to provide a name to a medical condition that someone is having signs and symptoms of. Depending on the type of testing done, the results of may also be beneficial for other family members to determine if they are also at risk.
- An example of this may be a woman who has previously had breast cancer who would like to have genetic testing to find out if she is at increased risk for other types of cancer, but also to provide information to her children, who may also be at an increased risk for cancer.
In other situations, the potential information that comes with genetic testing may be information that someone does not want to know.
- An example of this may be a man who has a family history of Huntington’s disease, a rare condition that affects everything from someone’s mental function to behavior to their ability to get around on their own. Huntington’s is an autosomal dominant genetic condition, so if his father had it, there is a 50% chance that he will also have it. Testing positive for Huntington’s will let someone know that they will go on to develop the disease, but there are no treatments that are currently available. Some people who are at risk for conditions like Huntington’s would not like to know if they have it ahead of time because it would cause them much stress and anxiety, and they don’t feel like there’s anything productive they can do with that information. Other people would like to know so they can do things to prepare.
There is no right or wrong answer when it comes to whether or not to pursue genetic testing; it is a personal choice. Genetic counselors are healthcare professionals that have special training in genetics. They can help to make sure that you have all of the information you need to make an informed decision that is right for your personal beliefs, and are also there to help support you in that decision.
With genetic testing, there are generally three different results that we can get: a positive, a negative, or a variant of uncertain significance (VUS).
Positive
A positive result means that a harmful change (called a pathogenic variant) was found in a gene that causes that gene to not work properly. The health issues that are cause by a pathogenic variant determine what gene it is found in. For example, pathogenic variants in the BRCA1 or BRCA2 genes can cause an increased risk for hereditary breast and ovarian cancer, while pathogenic variants in genes that help build your red blood cells can cause inherited types of anemia. A positive result can also mean that the lab found a likely pathogenic variant, which is a change that probably makes the gene not work correctly, but more research is needed to say for sure.
Pathogenic variants can also travel in families in different inheritance patterns, so finding out which gene is causing a particular health problem in a family may help to determine what other family members may be at risk and should also consider genetic testing.
A specialist in genetics, such as a genetic counselor, can help you understand how a positive genetic testing result may affect your health, and how it may affect the health of family members.
Negative
A negative result means that no harmful changes were found in any of the genes that were looked at. What a negative genetic testing result means depends on what the test was for, as well as the person’s medical and family history. Negative test results may also sometimes list gene changes that are benign (not harmful) or likely benign (probably does not change how the gene works, but more research is needed to say for sure).
If there has previously been a pathogenic variant that was identified in the family that someone was tested for (familial pathogenic variant), then we would call the result a true negative. That means that we know what the specific pathogenic variant is that caused the medical condition in their family, and that they did not inherit it.
- An example of this is if your father is found to carry a pathogenic variant that is linked to an increased risk for colon cancer, and you test negative for that variant, then we would call that a true negative. Because we know what contributed to the colon cancer in your family and that you did not inherit it, you would likely have the same risk for colon cancer as anyone else in the general population.
If there has not previously been a pathogenic variant identified in the family, we would call the result an uninformative negative. That means that there is a significantly decreased, but not zero, chance that there is not a pathogenic variant in the genes that were tested. The reason that we cannot say there is zero chance for a pathogenic variant in these genes is that our technology is not perfect, and our understanding of genetics is constantly evolving. A negative genetic testing result can mean different things depending on what the testing is looking for, your medical history, and your family history.
A negative genetic testing result should always be put into context given the medical and family history to help make the most sense out of it. A healthcare professional with a background in genetics, such as a genetic counselor, can help to do that.
Variant of Uncertain Significance (VUS)
A variant of uncertain significance (VUS) means that a change was found in one of the genes that the lab was looking at, but there is not enough information on that specific change to know for sure whether it is just a normal variation (we all have many of these), or whether it is a change that damages the gene and causes a genetic condition or an increased risk for a genetic condition.
When the lab finds a VUS, they continue to do more research to try to determine whether that specific change is harmful or not. It can sometimes take months or even years for them to figure that out. Most VUS results are ultimately found by the labs to be not be harmful, so many times we will treat this as a tentative negative until we get more information about them.
Getting a VUS result can sometimes lead to more feelings of uncertainty, stress, and anxiety. A healthcare professional with a background in genetics, such as a genetic counselor, can help to put a VUS result into context given your personal medical and family history.
Disorders & Conditions
There are a variety of genetic disorders and conditions that exist. The information provided below will help to provide a basic background of common disorders and conditions, and can be a resource in answering questions.
Alzheimer’s disease (AD) is a neurodegenerative disease that causes dementia. There are many different causes of dementia, with AD being the most common (60-80% of all dementia).
AD is caused by changes to the brain that lead to damage and breakdown of the nerve cells (called neurons). Accumulation of specific proteins (beta-amyloid and tau) in the brain interfere with how the neurons function and communication, and can lead to the death of neurons. The presence of these proteins are also thought to cause an inflammatory response which contributes to this damage, and can ultimately lead to the brain wasting away. AD also impacts the brain’s ability to use glucose, which is a primary source of energy for the brain’s functions. Other types of changes in the brain have also been found, however there are still significant limitations in our understanding of the brain changes and progression of disease seen with AD. In most cases, these changes to the brain start years to decades before an individual may start showing symptoms.
There are different phases of AD, including:
- pre-clinical phase: this is when someone has brain changes consistent with AD but are not showing symptoms yet.
- mild cognitive impairment phase: this is when someone has brain changes consistent with AD and have started to develop some memory and/or other issues with cognitive function. These difficulties may be noticeable to the individual and their family, but may not be detected by others.
- dementia phase: this is when someone has brain changes consistent with AD and have noticeable cognitive changes. An individual will often progress from mild to severe dementia over a period of time that differs for everyone. The degree of dementia often represents the amount of changes to the brain.
The length of time someone spends in each of these phases is variable. When an individual is suspected to have AD, the diagnosis is made based on a detailed review of the medical and family history, cognitive tests, ruling out other causes of dementia, and for some may involve specific lab tests and neuroimaging studies. These evaluations are done to look at and document cognitive, psychiatric, and other types of changes, and to rule out other potential causes that may have different treatments.
Categories of Alzheimer's
AD is often separated into two categories – late-onset AD and early-onset AD. Late-onset AD (LOAD) refers to those that develop dementia due to AD when they are 65 or older, and accounts for about 95% of AD. Early-onset AD (EOAD) refers to those that develop dementia due to AD before the age of 65, and accounts for less than 5% of AD. Both of these can present as sporadic cases (no family history) or familial cases (someone else in the family also has it). While most sporadic cases are late-onset AD and most familial cases are early-onset AD, there can be sporadic early-onset AD and familial late-onset AD. More than 75% of AD is considered sporadic, and about 15-25% of all AD is familial. Other causes of AD include Down syndrome, which accounts for about 1% of all AD.
Late-onset Alzheimer’s Disease (LOAD)
The primary risk factors for late-onset AD are age and genetics. Overall, about 10-12% of those in the general population who are 65 and older have dementia due to AD, and more than 30% develop it by age 85. There are multiple genes that have been identified that increase the risk of AD. These genes are often called “susceptibility genes,” which means changes in these genes may increase the risk to develop AD, but they do not cause AD on their own. The gene that has thus far been most strongly linked with AD is the APOE gene. The estimated heritability (amount of late-onset AD due to genetic susceptibility factors) is 60-80%.
Other risk factors that impact an individual’s chance of developing AD include those that can be changed. Some factors that increase the risk for dementia that can be changed include:
- less education
- hypertension (or high blood pressure)
- hearing impairment
- smoking
- obesity
- depression
- physical inactivity
- diabetes
- low levels of social contact
- excessive alcohol consumption
- traumatic brain injuries
- air pollution
Modifying these risks may help prevent or delay 40% of all dementias. In addition to the multiple genetic and environmental factors that can influence an individual’s chance to develop AD, a family history of AD may also increase the risk. However, an individual does not have to have a family history of AD in order to develop it, and similarly a positive family history does not guarantee an individual will develop AD.
Early-onset Alzheimer’s Disease (EOAD)
Early-onset AD is characterized by being diagnosed before the age of 65. There is also often a positive family history of AD, and many individuals with EOAD will have an underlying genetic cause for their early onset AD (90-100% heritability). The most well-known causes of early-onset AD are those inherited in an autosomal dominant pattern. About 1% of all AD and 10-15% of early-onset AD is caused by autosomal dominant AD.
Autosomal dominant (AD) is suspected when there are multiple individuals in the family across multiple generations who have with AD. The majority of autosomal dominant AD is due to pathogenic (harmful) variants in the genes presenilin 1 (PSEN1, 20-80%), amyloid-beta precursor (APP, 10-15%), and presenilin 2 (PSEN2, <5%). There are cases of early-onset AD with no family history (sporadic) but are caused by a pathogenic variant in one of these genes; this is called a de novo (or new) variant in the gene. These genes account for only about 5-10% of all early-onset AD. The remaining of early-onset AD are due to other or unknown causes. This means for the majority of those with early-onset AD, the specific cause may be not yet identified, or may be due to multiple genetic or other factors.
Genetic Testing for Alzheimer's
Genetic testing may be beneficial for those with a personal or family history of AD, particularly in those with early-onset AD and a family history of AD. For early-onset AD, genetic testing often involves sequencing and deletion/duplication analysis of the genes APP, PSEN1, and PSEN2. A positive result would indicate a pathogenic variant in one of these genes has been found and is the suspected cause of AD in that individual or family. A negative result would only indicate that the cause of AD is likely not due to one of these three genes. A negative result is common, as the cause of the majority of both early- and late-onset AD is unknown. While it is generally recommended that testing be done on an individual with AD, this is not always an option. Genetic counseling prior to and after testing is recommended.
Currently, genetic testing of the APOE gene is not routinely done outside of research studies because the predictive value (our ability to use the information from the testing to predict risks for someone) are variable and limited. Many people who have APOE test results that say they have a higher risk for AD will not every develop AD. Similarly, many people who APOE test results say they have a lower risk for AD may actually develop AD. For those who do undergo genetic testing and receive a result regarding their APOE genes, genetic counseling is recommended.
Treatment/Management for Alzheimer's
There is no known treatment for AD. There are medications and therapies that may help some individuals with their cognitive functions, but none are able to slow or stop the progression of the disease. Medical management is focused on providing proper care through appropriate treatments, therapies, activities, or support services, managing other health conditions the individual may have, and providing support to the caregiver.
Epilepsy is a group of neurological disorders in which individuals are at risk for recurrent/repeating seizures. The diagnosis of epilepsy is made when someone has two or more unprovoked seizures during their lifetime. In the general population, about 1 in 10 (10%) people will have one seizure during their lifetime, and about 3% will be diagnosed with epilepsy.
Causes of Epilepsy
There are a number of different causes for epilepsy. The majority of epilepsy is caused by a combination of genetic and non-genetic factors, with most related to infection, stroke, head trauma, brain tumor, or a brain malformation. Factors that impact whether an individual has an underlying genetic cause to their epilepsy include:
- the type of seizure (partial, generalized, or other)
- age of onset
- family history
- other health issues, such as intellectual disability, autism spectrum disorder, or birth defects.
An underlying genetic cause can be identified in 30-50% of those with epilepsy. The genetic causes may include chromosomal abnormalities or single gene disorders. The different causes may be de novo (new) or inherited in an autosomal dominant, autosomal recessive, X-linked, or mitochondrial pattern.
What type of genetic testing may be recommended?
For those with epilepsy, particularly with those who have symptoms in childhood, there are multiple types of genetic tests that may be considered. Recommendations may differ between patients depending on whether they have other health issues. Testing may include biochemical screening, chromosome microarray, single gene tests, multigene panels, or whole exome sequencing.
For those with other health conditions such as intellectual disability, autism spectrum disorder, or birth defects, the chromosome microarray may be considered as an initial test. The chromosome microarray is used to identify small missing or extra pieces of chromosome material, and can identify or rule out several hundred known genetic syndromes. This testing has a detection rate (the chance for the testing to come back positive) of about 20-30% in general. For all individuals with epilepsy the detection rate is less than 5%.
Further testing that may be considered includes single gene testing, multigene panels, or whole exome sequencing. About 15-40% of individuals with epilepsy are found to have a single gene disorder on one of these tests.
Genetic testing should be considered for those with epilepsy because in some cases finding the underlying genetic cause of someone’s epilepsy may change what is recommended for treatment. For instance, for some genetic forms of epilepsy there may be specific medications that should or should not be taken, or other treatments like dietary management that can be helpful.
Fabry disease, also called alpha-galactosidase-A deficiency, is classified as a lipid storage disorder. This means that people with Fabry disease are unable to metabolize (breakdown) lipids (fats) properly. Because this process occurs within the lysosomes of our cells (which remove waste from the cell), it is also called a lysosomal storage disorder. This leads to a build-up of lipids in the body, which can cause pain in the hands and feet, areas of small dark red spots called angiokeratomas, not being able to sweat properly (hypohydrosis), a clouding of the cornea (front part of the eye), and hearing loss. In addition, the lipid accumulation can lead to an increased risk of a heart attack or stroke, an enlarged heart (cardiomyopathy), and/or kidney disease (renal insufficiency or renal failure or end stage renal disease).
There are two more mild types of Fabry disease, called the ‘cardiac variant’ and the ‘renal variant’. For both, individuals often will not show symptoms or have significant health issues until later in life. People with the cardiac variant may have hypertrophic cardiomyopathy, cardiac arrhythmia, heart failure, or other heart problems. People with the renal variant of Fabry disease may have varying degrees of kidney disease, and may or may not have issues with other organs in the body.
Causes of Fabry Disease
We have over 20,000 different genes in the body. These genes are like instruction manuals for how to build a protein, and each protein has an important function that helps to keep our body working how it should. The GLA gene makes a protein called alpha-galactosidase-A (GLA). The GLA protein helps to break down a fatty substance in our cells called globotriaosylceramide (GTS). By breaking down GTS in the cells, the GLA protein prevents it from building up. If someone has a harmful change (called a pathogenic variant) in their GLA gene, then their body is not going to make as much GLA protein as it should. If the body does not have enough GLA protein, then the cells can not break down the GTS fats like they should, which leads to it building up in the cells. This is what leads to the signs and symptoms we associate with Fabry disease.
The GLA gene is located on the X chromosome, so Fabry disease is inherited in an X-linked pattern. Males only have one copy of their X chromosome, and therefore only one copy of all the genes that are on the X chromosome. Females have two X chromosomes, so they have two copies of all the genes on the X chromosome. This means that males with Fabry disease will produce little to no GLA protein, while females may produce some due to their second working copy of the gene. Because of this difference, males are often more severely affected and will often develop health issues at younger ages in comparison to females.
It is estimated that approximately 1 in 40,000 to 1 in 60,000 males have Fabry disease. Women can also be affected with Fabry disease but because the symptoms are generally much less severe, it is unknown how common Fabry disease is in women.
Genetic Testing for Fabry disease
Genetic testing for pathogenic variants in GLA is available. There are several different ways to approach to testing depending on the medical and family history, and any prior testing that may have been done. Different approaches include:
- Single site analysis: Testing specific to a known pathogenic variant in the family
- Full gene sequencing and rearrangement analysis: Comprehensive testing to search for all currently detectable pathogenic variants in the GLA gene.
- Gene panels: Newer, more broadly based gene tests that would include not only the GLA gene, but other genes known or suspected to be associated with the patient’s medical concerns
Diagnosing Fabry Disease
Fabry disease can be diagnosed either through genetic testing, or through a blood test that can measure the amount of GLA protein. There are also some red flags that could increase the chance for Fabry disease in a family:
- Periods of severe pain in the arms and/or legs
- Areas of small dark red spots called angiokeratomas
- Differences in the ability to sweat (most commonly not sweating enough or at all)
- Clouding in the front part of the eye (cornea)
- An incident of stroke or a specific type of heart disease called left ventricular hypertrophy that is seemingly unexplained
- Reduced kidney functioning with no known cause
Treatment/Management for Fabry Disease
Once the diagnosis of Fabry disease is confirmed, screening may be recommended to evaluate for related health issues. This may include heart, kidney, hearing, eye, neurological, vascular, and other evaluations. To keep an eye on the progression of disease, people with Fabry disease will also need to have routine screening of their kidneys, heart, and hearing (and possibly other screening depending on their personal health concerns).
Fabry disease can also be treated by the use of enzyme replacement therapy (ERT), which involves replacing the missing GLA protein (an enzyme is a specific type of protein). This is done through intravenous (IV) infusion. In the United States there are two approved forms of ERT available, called Replagal® and Fabrazyme®.
These are generalized recommendations for Fabry disease. It is important for affected individuals to work with a medical provider who is familiar with Fabry disease to come up with a medical management plan that is best for them.
Familial hypercholesterolemia (FH) is an inherited condition where the body is not able to get rid of extra cholesterol, causing it to build up in the bloodstream and other parts of the body. This buildup of cholesterol can narrow the heart valves and arteries, which increases the chance for cardiovascular disease such as stroke, heart attack, and heart disease. These conditions are also often diagnosed at younger ages in people with FH than would normally be expected. As many as 30-50% of people with FH may also develop small yellowish patches of cholesterol buildup called xanthomas. Xanthomas usually occur around the eyelids and in the tendons in the feet, hands, knees, and elbows, and may get smaller with treatment. About half of all men with FH will have a health issue with their heart or their arteries by the age of 50, while 85% will have a heart attack by the age of 60. In women with FH, about 30% will experience a health issue with their heart or arteries by the age of 60.
Cholesterol is needed by the body to work properly, and it helps the body to make hormones, vitamin D, and certain substances that help with food digestion. There are two main types of cholesterol, called HDL and LDL. LDL works to deliver cholesterol to your arteries, while HDL works to move cholesterol to your liver. The liver removes extra cholesterol from the body, preventing it from building up. LDL cholesterol is the type that most doctors are talking about when they tell someone they have high levels of the ‘bad’ cholesterol.
For most people with high LDL cholesterol, their diet or lifestyle causes an increase in the amount of LDL cholesterol in their body, and the liver can not get rid of the extra cholesterol fast enough. For people who have FH, their high levels of LDL cholesterol are not directly because of their diet or lifestyle (although those also do play an important part), but because their liver is not able to remove the extra LDL cholesterol.
Causes of Familial Hypercholesterolemia
Approximately 70-95% of cases of FH are caused by harmful changes (called pathogenic variants) in one of three genes: LDLR (60-80% of all cases of FH), APOB (1-5% of all cases of FH), and PCSK9 (0-3% of all cases of FH). The remaining cases have an unknown cause, but may be due to pathogenic variants in other genes that we do not know about yet.
Recent studies have estimated that as many as 1 in 200 (0.5%) people may have FH, and that 2-3% of all heart attacks that happen before the age of 60 may be due to FH. Between 1 in 160,000 and 1 in 250,000 people inherit a FH gene from each parent that has a pathogenic variant in it (called homozygous). Most people who have two non working FH genes have severe coronary artery disease by their mid-20s, and the risk for sudden death or bypass surgery is high during the teenage years. People who are homozygous may also develop xanthomas in between their fingers.
Diagnosing Familial Hypercholesterolemia
While genetic testing for FH can be helpful to establish a diagnosis, medical providers may use other pieces of information from lab tests, a physical exam, and the family history to determine if someone is at a higher chance to have FH. Some red flags that can increase the chance for FH in a family include:
- Extremely high cholesterol
- In adults, this would be an LDL cholesterol level of over 190 mg/dL, or a total cholesterol level of over 310 mg/dL.
- In children, this would be an LDL cholesterol level of over 130 mg/dL, or a total cholesterol level of over 230 mg/dL.
- A personal or family history of early-onset (generally thought to be before the age of 45 in men and before the age of 50 in women) coronary artery disease or cardiovascular disease (including angina, heart attack, and vascular disease)
- Physical exam that finds xanthomas at any age or corneal arcus (a gray or white arc or ring around the colored part of the eye) before the age of 45
It is thought that most people (up to 90%) who have FH have not been properly diagnosed. If you or members of your family have any history that is concerning for the signs and symptoms of FH, you should speak with a specialist, such as a cardiovascular specialist or genetic counselor, to get more information.
Treatment/Management for Familial hypercholesterolemia
Treatment for FH can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with FH. Some options for medical management can include regular blood work, medications, dietary recommendations (reduced saturated fat and increased soluble fiber), and imaging (including echocardiograms and CT angiograms).
People with FH should avoid risk factors for cardiovascular and coronary artery disease, such as smoking, high fat and high cholesterol diet, obesity, and high blood pressure. Regular physical activity and following dietary recommendations can help with weight control as well as overall cardiovascular health.
Leukodystrophies are a group of genetic disorders that affect the white matter of the central nervous system, which consists of your brain and spinal cord. The white matter is made up of a complex network of axons, which are how nerve cells communicate with each other. Axons are typically insulated by a substance called myelin that helps move these communications along. Leukodystrophies refer to the abnormal development of the white matter, or the progressive breakdown of the white matter. This abnormal development or breakdown occurs due to an underlying genetic condition that impacts the axon, myelin, or other parts of the central nervous system that are important for how the axon or myelin function.
There are several dozen different types of leukodystrophies (over 50 total). Some of the more common types include:
- Metachromatic leukodystrophy (MLD)
- X-linked adrenoleukodystrophy (ALD)
- Pelizaeus-Merzbacher disease (PMD)
- Krabbe disease
- Canavan disease
- Alexander disease
Anywhere from 1 in 6,000-7,000 to 1 in 100,000 individuals are affected with a leukodystrophy.
Because the onset, severity, and progression of different leukodystrophies is extremely variable, it is difficult to provide generalized statements about the signs and symptoms that people with leukodystrophies may experience. Some types of leukodystrophy have a predictable disease progression, while other types are highly variable. For instance, while many symptoms associated with leukodystrophies get worse over time, the rate of this progression can vary. Some forms may have rapid decline and be life shortening, while other types have a more gradual and benign progression. There are some types, although less common, that can show improvement or stabilization over time. Others can appear to be stable, only to have a rapid decline after a triggering event.
The age of onset also varies amongst the different types of leukodystrophy, and sometimes even within the same type of leukodystrophy. Individuals may start to show symptoms anywhere ranging from birth through adulthood. The onset is often broken down into categories:
- Neonatal
- Infantile
- Late infantile
- Juvenile
- Adolescence
- Adulthood
The earlier onset is often associated with more severe and rapid progression and decline, but this too can be variable.
Diagnosing Leukodystrophy
The diagnosis of leukodystrophy and identifying the specific type of leukodystrophy someone has is based on age of onset of symptoms, brain MRI patterns, detailed examination for both neurological and non-neurological symptoms or features, and biochemical and genetic tests. Due to the use of multi-gene panels and whole exome and genome sequencing, more people are able to receive a diagnosis of their specific type of leukodystrophy compared to the past.
Some neurological symptoms that can be seen in leukodystrophy include:
- Developmental delays or intellectual disability
- Motor impairment, such as delays in reaching motor developmental milestones, onset of abnormal movements or movement disorders, abnormal gait, or changes in muscle tone
- Psychiatric or behavioral disorders; these types of symptoms are usually some of the first that can be seen
- Dysautonomia
- Seizures
- Peripheral neuropathy
Non-neurological symptoms can also be variable, and can affect many of the other body systems. The specific non-neurological symptoms can sometimes be used to help determine the type of leukodystrophy that someone has. However, because the signs and symptoms of the many different types of leukodystrophies overlap, further testing is often needed.
A brain MRI is one of the most helpful tools to diagnose what specific type of leukodystrophy someone has. The type and pattern of lesions that are seen on the brain MRI can help determine if someone has a genetic white matter abnormality (leukodystrophy) or an acquired (non-genetic) white matter abnormality. This information can be used to help determine specific types of leukodystrophy that may be more likely.
The combination of clinical features, age of onset, and brain MRI findings may allow for a more targeted testing approach with metabolic or genetic testing. If the combination of these things do not point to one specific type of leukodystrophy, broader testing with multi-gene panels or whole exome and genome sequencing could be helpful. It is important to remember that not all individuals with a clinical diagnosis of leukodystrophy will have an identifiable genetic cause. Through the use of multi-gene panels and whole exome and genome sequencing, as many as 70-80% people are able to receive a specific diagnosis. This means for about 20-30% of individuals with leukodystrophy, a genetic cause is not able to be found at this time. As our understanding of genetics continues to progress, this percent will likely continue to decrease.
Treatment/Management for Leukodystrophy
For most leukodystrophies there is no known treatment, and medical management is focused on treating the symptoms (both neurological and non-neurological). For some types of leukodystrophy there is treatment available; for instance, hematopoietic stem cell transplantation (HSCT) is an available option for several types such as metachromatic leukodystrophy, Krabbe disease, and cerebral adrenoleukodystrophy. Other forms of treatments being looked at for leukodystrophies include gene therapy, antisense oligonucleotide therapy, and other targeted approaches. However, as with HSCT, the timing of these treatments is crucial. If an individual is treated after development of white matter disease, the treatment may not be as effective and may only stabilize the patient’s symptoms.
Malignant hyperthermia (MH) is a genetic condition where individuals are more likely to have severe reactions to specific types of medications. For this reason, it is also called “malignant hyperthermia susceptibility” (MHS). The types of medications people may have reactions to include certain gas anesthetics (used to block pain) and the muscle relaxant succinylcholine (used for temporary paralysis during surgery or other procedures). If exposed to one of these medications, individuals with MHS are at risk to develop a variety of symptoms that include increased levels of acid in the body (which can cause increased or rapid heart rate, or possibly cardiac arrest), increased levels of carbon dioxide in the body (which can cause abnormal breathing), muscle rigidity, breakdown of muscle fibers with leakage into the blood and urine (rhabdomyolysis, which can be harmful to the kidneys), and hyperthermia (abnormally high body temperature). Those with MHS may also show symptoms after exercise and/or heat stress. Without proper treatment MHS can be life-threatening.
Causes of Malignant Hyperthermia Susceptibility
About 70-80% of MHS is caused by harmful changes (called pathogenic variants) in the gene RYR1, and about 1% is caused by pathogenic variants in the CACNA1S gene. The remaining 20-30% is either caused by unknown genetic causes, or the MHS is a part of a specific genetic condition caused by pathogenic variants in other genes. For example, many neuromuscular disorders increase the risk for MH.
The STAC3 gene has also been shown to cause MHS. Individuals reported with MHS due to pathogenic variants in the gene STAC3 are often of Native American descent, and also have a condition called Native American Myopathy. This condition is inherited in an autosomal recessive pattern, so a pathogenic variant must be present in both copies of the gene.
Because individuals may not know that they have MHS unless they are exposed to one of the medications that can cause a reaction, estimating how common it is can be difficult. Current measurements include looking at the number of people who have had an issue during anesthesia with one of these medications, and were then found to have MHS. Those numbers show that approximately 1 in 5000 to 1 in 50,000 people are suspected to have MHS.
Diagnosing Malignant Hyperthermia Susceptibility
While genetic testing for MHS can be helpful to establish a diagnosis, medical providers may use another test, called a muscle contracture test (also called caffeine halothane contracture test, CHCT). This testing involves looking at a skeletal muscle biopsy (piece of muscle that is surgically removed) after being exposed to substances such as caffeine or halothane (called ryanodine receptor agonists). Individuals who have MHS will have abnormally high levels of force in the contraction of their muscle biopsy sample when exposed to one of these substances.
CHCT may be considered in people who have a positive family history of MHS, a previous suspected episode of MHS, a history of severe muscle rigidity when exposed to one of the triggering substances, a history of moderate or mild muscle rigidity with rhabdomyolysis, history of unexplained rhabdomyolysis during or after surgery or after a negative rhabdomyolysis workup, or when there are other signs that increase the chance for MHS.
Unfortunately because there are a very limited amount of medical centers in the country that do CHCT testing, a majority of people will have to travel to one of these centers in order to have the testing done.
Treatment/Management for Malignant Hyperthermia Susceptibility
The main thing that is important for individuals with MHS to do for medical management is to avoid the gas anesthesthetic and succinylcholine that can cause a reaction. If someone who has MHS is having general anesthesia that goes on longer than 30 minutes, it is important to have their temperature monitored. It is also important for people who have a family history of MHS (and may also be at risk for it themselves) to let their medical providers know and to take the recommended precautions. There may also be other blood or urine tests that would be helpful to do when someone has just been diagnosed with MHS.
If someone is not known to have MHS but begins having a reaction under anesthesia, it is important for the medical team to be able to identify the signs early and begin treatment. If the episode is not caught early, more long-term damage to the patient’s health can result.
Neurometabolic conditions refer to metabolic disorders that impact brain development or function. Individuals with these disorders are either not able to produce certain enzymes, proteins, and/or other substances needed by the brain, or they are unable to break down these substances, which can cause them to build up and can be toxic to the brain.
As a whole, neurometabolic disorders range both in age of onset and severity, even within the same specific disorder. People with neurometabolic disorders can show symptoms anywhere from birth all the way through adulthood. Individuals who have symptoms at birth or in infancy tend to be more severely affected, but there is wide variability in the number and severity of symptoms. Some symptoms that can be seen with neurometabolic disorders include:
- abnormal brain structures
- seizures
- poor development (also called neurodevelopmental delay)
- neurological regression (e.g. loss of skills)
- movement disorders
- abnormal behaviors
- sleep disorders
- psychiatric disorders
- other body system involvement (also called multi-systemic involvement).
Diagnosing Neurometabolic Disorders
Diagnosis of a neurometabolic disorder is similar to how metabolic disorders in general are diagnosed, which is:
- through careful evaluation of an individual’s medical and family history
- physical examination
- measurement of specific levels of certain substances in the blood, urine, cerebrospinal fluid (CSF), or other body tissues (e.g. skin or muscle)
- neuroimaging (MRI, functional MRI, MRS)
- other studies, and/or genetic testing.
Treatment/Management for Neurometabolic Disorders
While many neurometabolic disorders do not have known treatments, early diagnosis is crucial so those with an available treatment can be started early. If these conditions lead to any sort of brain damage, the brain is often not able to regain full function. Thus, treatment is focused on reducing and eliminating the buildup of the toxic substances in the brain to prevent the damage from occurring in the first place. This may be done through a specialized diet, taking supplements, or through replacing the missing enzymes. Gene therapy is also being assessed for a handful of these disorders through clinical trials. Starting disease-specific treatments during this crucial early period is dependent on having an accurate diagnosis.
Examples of neurometabolic disorders include:
- Amino acid disorders (e.g. phenylketonuria, PKU)
- Peroxisomal disorders (e.g., adrenoleukodystrophy, ALD or Zellweger or Neuronal Ceroid-lipofuscinosis, NCL)
- Lysosomal storage disorders (e.g. Tay-Sachs disease or mucopolysaccharidoses, MPS)
- Mitochondrial disorders (e.g. Leigh syndrome)
- Neurotransmitter disorders (e.g. PTPS deficiency)
- Other (e.g. GLUT1 deficiency)
Neurodegenerative conditions refer to a group of disorders that impact the way a person’s brain and nerves function. Individuals with these disorders have nerve cells, called neurons, in the brain and throughout the body that lose function over time, and eventually these neurons will lose all function and die. Neurons don’t replicate or repair themselves, so our bodies do not have a way to make more after these cells die. When these neurons die, it affects the brain’s ability to work and communicate correctly with the rest of the body.
Some neurodegenerative conditions we know are inherited and caused by changes in specific genes, while other conditions we are still learning about. Neurodegenerative disorders range in symptoms and severity. Some symptoms that can be seen with neurodegenerative disorders include:
- Involuntary movement
- Problems with memory or thinking
- Stumbling
- Shaking
- Mood changes
Diagnosing Neurodegenerative Disorders
Diagnosis of a neurodegenerative disorder can include:
- Careful evaluation of an individual’s medical and family history
- Physical examination
- Neuroimaging (MRI, functional MRI, MRS)
- Other studies, and/or genetic testing.
Treatment/Management for Neurodegenerative Disorders
There is currently no known cure for neurodegenerative disorders, but clinical trials are evaluating potential treatments.
Examples of neurodegenerative disorders include:
- Huntington’s Disease (HD)
- Amyotrophic Lateral Sclerosis (ALS)
- Frontotemporal dementia (FTD)
- Alzheimer’s Disease
- Parkinson’s Disease
- Leukodystrophies
Ornithine transcarbamylase (OTC) deficiency is classified as a urea cycle disorder. The urea cycle is a process in which the body removes ammonia (a waste product from the breakdown of proteins) from the body. This process most commonly occurs in the liver. People with a urea cycle disorder have difficulties breaking down and getting rid of ammonia, which can cause an accumulation of ammonia in the body that can be toxic, particularly to the neurological system.
This toxicity to the body is what will cause the symptoms seen in OTC deficiency. Symptoms can begin as early as the newborn period, or as late as adulthood. Some of the health issues that OTC deficiency can lead to include periods of vomiting, lethargy, tachypnea (hyperventilation), failure to thrive, or acute fatty liver. Between episodes they can have anorexia, nausea, and protein intolerance. Other complications of OTC deficiency may include seizures, behavioral problems, developmental delays, cognitive impairment, and other types of neurological concerns. In some cases, if not recognized early or left untreated, OTC deficiency can lead to coma and even death.
Causes of OTC Deficiency
We have over 20,000 different genes in the body. These genes are like instruction manuals for how to build a protein, and each protein has an important function that helps to keep our body working how it should. The OTC gene makes a protein called ornithine transcarbamylase (OTC). The OTC protein is active in helping with urea cycle in the liver to prevent ammonia from building up, which can be toxic to the nervous system. If someone has a harmful change (called a pathogenic variant) in their OTC gene, then their body is not going to make as much OTC protein as it should. If the body does not have enough OTC protein, then the urea cycle cannot work like it should, which leads to a toxic buildup of ammonia in the body. This is what leads to the signs and symptoms we associate with OTC deficiency.
The OTC gene is located on the X chromosome, so OTC deficiency is inherited in an X-linked pattern. Males only have one copy of their X chromosome, and therefore only one copy of all the genes that are on the X chromosome. Females have two X chromosomes, so they have two copies of all the genes on the X chromosome. This means that males with OTC deficiency will produce little to no OTC protein, while females may produce some due to their second working copy of the gene. Because of this difference, males are often more severely affected and will often develop health issues at younger ages in comparison to females.
It is estimated that approximately 1 in 14,000 to 1 in 77,000 people have OTC deficiency. It may be more common because some people may have mild symptoms and not show any symptoms until much later in life.
Genetic Testing for OTC Deficiency
Genetic testing for pathogenic variants in OTC is available. There are several different ways to approach to testing depending on the medical and family history, and any prior testing that may have been done. Different approaches include:
- Single site analysis: Testing specific to a known pathogenic variant in the family
- Full gene sequencing and rearrangement analysis: Comprehensive testing to search for all currently detectable pathogenic variants in the OTC gene.
- Gene panels: Newer, more broadly based gene tests that would include not only the OTC gene, but other genes known or suspected to be associated with the patient’s medical concerns
Diagnosing OTC Deficiency
The diagnosis of OTC deficiency is often made following an episode or medical history that is concerning for OTC deficiency, or due to a positive family history. OTC deficiency is often diagnosed through measurements of levels in the blood and urine in combination with either genetic testing or measurement of enzyme activity in the skin or liver (both would require a biopsy to be done). OTC deficiency is also detected through newborn screening in some states.
Treatment/Management for OTC Deficiency
Management of OTC deficiency is focused on preventing episodes of high ammonia levels (which can lead to a crisis and the symptoms described above). Affected individuals may require a protein-restricted diet, the use of nitrogen scavengers (medications that help remove ammonia), and other medical supplements. Some people with OTC deficiency may eventually require a liver transplant if the condition becomes difficult to manage. This is particularly true for those who start showing symptoms in the newborn period.
Precautions are also taken to help reduce the risk for an episode. This includes avoiding fasting, stress, and specific medications. For females, additional precautions may be put in place during pregnancy and delivery, due to the stress this can put on the body.
Wilson Disease is a genetic condition that causes the body to accumulate too much copper, which can affect the brain, liver, and eyes. The signs of Wilson disease often appear in the teenage years, and young adults with Wilson disease can develop problems with their nervous system, including issues with coordination, difficulty talking, impaired thinking, tremors, and psychiatric concerns (including mood disorders). Excess copper in the system can also damage the liver and can cause jaundice (yellowing of the skin and eyes) as well as fatigue.
Wilson disease can be diagnosed by measuring the amount of copper in the blood, urine, and liver. A characteristic sign of Wilson disease is a dark colored circle on the surface of the eye, called a Kayser Fleischer ring, caused by an accumulation of copper. The Kayser Fleischer ring does not hurt vision.
Causes of Wilson Disease
Wilson disease is caused by mutations (or changes) in the ATP7B gene. This gene’s function in the body is to help transport copper from the liver all around the body. It also is important to help the body get rid of excess copper. When this gene is not working properly and cannot get rid of extra copper in the body, it starts to build up and become toxic, which is what causes the signs and symptoms of Wilson disease.
Wilson disease is inherited in an autosomal recessive pattern. This means an individual who has Wilson disease has inherited two non-working copies of the ATP7B gene; the one they inherited from their mom is not working AND the one they inherited from their dad is not working. In the case of autosomal recessive conditions, if you inherit one working ATP7B gene from a parent and one non-working ATP7B gene from a parent, you are called a ‘carrier’ for Wilson disease. Carriers do not have Wilson disease, and typically do not have signs or features of Wilson disease.
Diagnosing Wilson Disease
Determining a diagnosis of Wilson disease can be complex and involve many different steps and types of testing, and should be done by a provider who is familiar with Wilson disease. The American Association for the Study of Liver Diseases (AASLD) published guidelines in 2008 that outline in detail the testing that is recommended to diagnose Wilson disease. Some findings that may raise the suspicion of Wilson disease are:
- Liver disease (recurrent jaundice, autoimmune-type hepatitis, or chronic liver disease)
- Neurological concerns (tremors, poor coordination, loss of fine-motor control, rigidity, gait disturbance, etc).
- Psychiatric concerns (depression, neurotic behaviors, personality disorganization, and intellectual deterioration.
- Kayser-Fleischer rings (copper deposits on the outside of the eye) occur in approximately 50-60% of people who have Wilson disease and liver disease, and in about 90% of people who have Wilson disease and neurologic or psychiatric concerns.
Genetic testing can also be helpful to establish the diagnosis of Wilson disease. Approaches may include:
- Single site analysis: Testing specific to a known mutation in the family
- Full gene sequencing and rearrangement analysis: Comprehensive testing to search for all currently detectable mutations in the ATP7B gene.
- Gene panels: A panel of genes that includes ATP7B and other genes that are suspected to be involved.
Treatment/Management for Wilson disease
Treatment options for individuals with Wilson disease may vary, but in general would start with copper chelating therapy, which include medications that increase the body’s ability to get rid of extra copper. Taking zinc supplements after this can help to interfere with the body’s ability to absorb excess copper, which can further reduce many of the symptoms that affect someone with Wilson disease. There is also lab work (blood and urine) that is recommended to do one to two times per year. Individuals with Wilson disease should try to avoid foods that have high levels of copper (liver, brain, chocolate, mushrooms, shellfish, and nuts), particularly at the start of treatment.
Cancer Predispositions
A variety of hereditary cancer predispositions exist. The information provided below will help to provide a basic background and answer common questions for individuals who have a personal and/or family history of cancer.
The two adrenal glands are located in the abdomen, one atop of each kidney, and produce a variety of hormones including adrenaline, aldosterone, and cortisol. Benign (noncancerous) adrenal tumors are relatively common in general population. However, a rare tumor of the adrenal gland called a pheochromocytoma (PCC) will secrete hormones into the bloodstream and can cause anxiety and uncontrollable high blood pressure.
A tumor that is similar to a PCC is called a paraganglioma (PGL). Unlike PCCs, PGLs are not limited to just the adrenal glands, and are commonly found in the head and neck region of the body. Adrenocortical carcinoma (ACC), or cancer of the adrenal cortex, is a rare type of adrenal cancer.
Compared with other types of tumors, PGLs and PCCs are very rare. However, the proportion of these rare tumors that are hereditary is much higher. As many as 25-40% of PCC and PGL tumors are considered to be due to a hereditary cause. Similarly, compared with other types of cancers, ACC is very rare. It is estimated that 10% of all cases of ACC are due to a hereditary cause. Children with ACC have a higher likelihood of having a hereditary cause than adults with ACC.
Because of the increased chance for these tumors and cancers to be hereditary, it is recommended that all patients consider genetic counseling regardless of what age they were diagnosed with PCC, PGL, or ACC, and whether or not they have a family history of cancer. There are currently at least a dozen genes that are known to contribute to PGL, PCC, and ACC, some of which overlap with other types of hereditary cancer syndromes. Some hereditary cancer syndromes that can include adrenal cancer are:
- Multiple Endocrine Neoplasia, type 2 (RET)
- Li-Fraumeni syndrome (TP53)
- Von-Hippel Lindau syndrome (VHL)
- Neurofibromatosis, type 1 (NF1)
- Carney complex (PRKAR1A)
- Hereditary paraganglioma-pheochromocytoma (SDHD, SDHAF2, SDHC, SDHB)
Hereditary breast and ovarian cancer (HBOC) is a genetic syndrome that increases the chances to develop certain types of cancer, including breast (female and male), ovarian, prostate, and pancreatic cancers, as well as melanoma in some families. Individuals with HBOC can also be diagnosed with cancer at younger ages than would normally be expected, with some people being diagnosed with cancer in their 20s.
Prostate cancer in men is usually not a fast-growing or rapidly spreading type of cancer, and many men with prostate cancer may not have to undergo extensive treatment. However, prostate cancer in men with HBOC is more likely to occur earlier than it does in men in the general population, and is more likely to metastasize (spread throughout the body) and pose a more significant threat to one’s health.
Approximately 1 in 500 to 1 in 1000 people in the general population have HBOC. It is estimated that HBOC contributes to approximately 5-10% of all breast cancers and 10-15% of all ovarian cancers. Approximately 6% of men with prostate cancer will have HBOC. HBOC is more common in some ethnic backgrounds, particularly the Ashkenazi Jewish population (where approximately 1 in 40 people of Ashkenazi Jewish ancestry are thought to have HBOC). The number of individuals who have HBOC may be higher than this because many people who have HBOC will never go on to develop cancer.
HBOC is caused by harmful changes (called pathogenic variants) in the BRCA1 and BRCA2 genes.
Diagnosing HBOC
A definitive diagnosis of HBOC can only be made by performing genetic testing to look for pathogenic variants in the BRCA1 and BRCA2 genes. The National Comprehensive Cancer Network (NCCN) is a group of medical professionals that regularly meet to look over any updates in research studies and determine recommendations for who should be considered at a higher risk for HBOC. Some patterns or red flags in a personal or family history that make it more likely that a family could have HBOC, including:
- A family history of ovarian cancer
- A family history of metastatic (cancer that has spread to other parts of the body) prostate cancer
- A family history of breast cancer diagnosed before age 50
- A family history of triple-negative (a specific type) breast cancer diagnosed before age 60
- A family history of three or more relatives (on the same side of the family), that have had breast, pancreatic, or prostate cancer
- Ashkenazi Jewish ancestry and a family history of breast, ovarian, pancreatic, or prostate cancer
If you or your family have a history of any of these types of cancer, it is recommended that you meet with a specialist, such as a genetic counselor, who can gather more information to help determine if you have a higher chance to have HBOC. It is important to note that not all people who have the above types of cancer in their family will have HBOC.
Treatment/Management for HBOC
If found to have HBOC, it is recommended to discuss your management plan with your healthcare team, and if available, to seek consultation through a specialized high-risk clinic. General recommendations are included here based on the updated guidelines of the NCCN, but may be tailored to your specific medical and family history.
Breast cancer screening for women with HBOC includes:
- Beginning at age 18: Breast self exam to facilitate awareness and familiarity with breast tissue
- Ages 25-29: Clinical breast exam every 6-12 months, and breast MRI every 12 months
- Ages 30-75: Clinical breast exam every 6-12 months, breast MRI every 12 months, and mammogram every 12 months
- Age 75+: Individualized management
Bilateral prophylactic (preventive) mastectomies or preventive medications are also available options for breast cancer risk reduction, and are a personal decision.
Women at increased risk for ovarian cancer due to HBOC are encouraged to consider prophylactic (preventative) removal of the ovaries and fallopian tubes (called a salpingo-oophorectomy) between age 35-40, or once they are finished having children. For women who do not elect preventive surgery, current ovarian cancer screening includes CA-125 blood levels and transvaginal ultrasound beginning at age 30-35, however this screening is not proven to be effective and is not recommended by many medical organizations.
It is recommended that men with HBOC talk with their doctor about screening options for prostate cancer at the age of 40. Currently, the most effective way to screen for prostate cancer is by doing a blood test to look for something called the prostate-specific antigen (PSA), which is a blood test that can be done yearly. It is important for men with HBOC to have an in-depth conversation with their medical provider about the risks, benefits, and limitations for PSA screening because there is the possibility for false positives (positive PSA result when someone doesn’t have prostate cancer) and false negatives (negative PSA result when someone actually has prostate cancer). Because of the possibility of false positives and negatives, some men may opt not to pursue PSA screening.
Juvenile polyposis syndrome (JPS) is a polyp predisposition syndrome, where there is a higher risk to develop polyps in your digestive tract running in the family. The name juvenile describes the specific types of polyps that develop, not the age when you develop them. This type starts with many small growths, or polyps, in your colon that can cause bleeding and anemia and may eventually develop into cancer if not removed. A specialized doctor called a gastroenterologist can go in and remove these growths before they turn into cancer.
Pathogenic (or harmful) variants in one of two genes cause JPS: BMPR1A and SMAD4. It is inherited in an autosomal dominant pattern, meaning that children of someone who carries a pathogenic variant each have a 50% risk to inherit the variant and associated cancer risks. Notably, women and men both have the BMPR1A and SMAD4 genes and have the same chances to inherit and pass down variants in these genes. Therefore, both sides of the family are important when assessing inherited risk. About 33% (or 1 in 3 people) with this condition will have a parent with polyps. The other 2 in 3 (or 67%) of people have no family history and will be the first ones in their family with this condition.
JPS is suspected in a person with any of the following:
- More than 5 juvenile polyps in the colon
- Multiple juvenile polyps anywhere else in the GI tract (such as stomach or small intestine)
- Juvenile polyps with a family history of someone else with juvenile polyps
Genetic Testing for JPS
Genetic testing is available for this condition. There are different ways to complete this testing. This can include:
- Single site analysis: Testing specific to a known pathogenic variant in the family
- Full gene sequencing and rearrangement analysis: Comprehensive testing to search for all currently detectable variants in the genes.
- Gene panels: Newer, more broadly based gene tests that would include not only the BMPR1A and SMAD4 genes, but other genes known or suspected to be associated with increased cancer risks.
Risks Associated with JPS
The risk that someone with JPS will get cancer over their lifetime is between 9 and 50%, including cancer of the colon, rectum, small intestine, and pancreas. The chance of stomach cancer is 21% in those who have gastric polyps. One study showed the risk for colon cancer may be as high as 68%.
There is also a risk for an increased number of polyps, from 4 or 5 to more than 100. These are not cancerous and many of them will stay benign, but they have the potential to develop into cancer. They can be anywhere in your digestive tract: stomach, small intestine, colon, or rectum.
The chance to develop colon cancer partly depends on if you get regular colonoscopies. A colonoscopy can reduce the risk of colon cancer by removing the polyps before they have a chance to develop into cancer.
Treatment/Management for JPS
- Routine colonoscopy (every 1 year if polyps are found, every 2-3 years if no polyps are found) starting at around age 15.
- Screening by complete blood count (CBC) and upper endoscopy starting at around age 15, or earlier if symptoms occur. Repeat every 1 year if polyps are found, and every 2-3 years if no polyps are found)
- When a large number of polyps are present, removal of all or part of the colon or stomach may be necessary.
- Treatment as needed for manifestations of HHT (see below).
Another thing to know about JPS is that people with a SMAD4 pathogenic gene variant have a higher chance of also having another condition called hereditary hemorrhagic telangiectasia (HHT). Our genes are very complex, and sometimes different changes in the same gene can cause different health issues. Some variants in the SMAD4 gene only cause JPS, while different variants in the same SMAD4 gene can cause JPS and HHT. This result may come up while doing testing for another, unrelated condition as part of general genetic screening. HHT can cause nose bleeds and an increased risk for aneurysms and arteriovenous malformations (groups of abnormal blood vessels connecting arteries and veins in the brain and lungs). There is treatment available for people with HHT.
Li-Fraumeni syndrome is caused by pathogenic (or harmful) variants in the TP53 gene. Individuals with Li-Fraumeni syndrome generally have a considerably high risk for several types of cancer (lifetime cumulative cancer risk is nearly 100%), can generally be diagnosed at significantly younger ages, and have an increased risk to get more than one type of cancer in their lifetime.
TP53 pathogenic variants are inherited in an autosomal dominant pattern, meaning that children of someone who carries a pathogenic variant each have a 50% risk to inherit the variant and associated cancer risks. Notably, women and men both have the TP53 gene and have the same chances to inherit and pass down variants in these genes. Therefore, both sides of the family are important when assessing inherited risk. However, the associated cancers and lifetime risks are different between men and women.
Cancers commonly associated with Li-Fraumeni syndrome include:
- Breast cancer
- Sarcoma (cancer of the soft tissue or bone)
- Adrenocortical cancer
- Brain cancer
- Colon cancer
- Lung cancer
- Leukemia
- Thyroid
- Others
Genetic Testing for TP53
Genetic testing for pathogenic variants in TP53 are currently available, but there are a few different ways to approach testing:
- Single site analysis: Testing specific to a known pathogenic variant in the family
- Full gene sequencing and rearrangement analysis: Comprehensive testing to search for all currently detectable variants in the gene
- Gene panels: Newer, more broadly based gene tests that would include not only the TP53 gene, but other genes known or suspected to be associated with increased cancer risks
Whether or not someone meets criteria for genetic testing for Li-Fraumeni syndrome can be determined by a couple different methods. The ‘classic’ Li-Fraumeni criteria include:
- A combination of an individual diagnosed with a sarcoma before age 45, AND
- A first-degree relative (sibling, parent, or child) diagnosed with cancer before age 45, AND
- An additional first- or second-degree (aunts/uncles, nieces/nephews, grandchildren) relative in the same lineage with cancer diagnosed before age 45, or who was diagnosed with a sarcoma at any age.
Another set of criteria, called Chompret criteria, is a little more complicated. It says that individuals should consider genetic testing for Li-Fraumeni syndrome:
- Individual diagnosed before age 46 with a Li-Fraumeni-associated tumor (soft-tissue sarcoma, osteosarcoma, central nervous system tumor, breast cancer, adrenocortical carcinoma, etc) AND at least one first- or second-degree relative with any of the Li-Fraumeni-associated tumor (other than breast cancer if the primary individual has breast cancer) before the age of 56 or with multiple primaries at any age, OR
- Individual with multiple tumors (except multiple breast tumors), two of which are one of the above associated tumor types, with the initial cancer being diagnosed before age 46, OR
- Individual with adrenocortical carcinoma, or choroid plexus carcinoma or rhabdomyosarcoma of embryonal anaplastic subtype, at any age of onset, regardless of the family history, OR
- Breast cancer before the age of 31
These criteria are very complex, which is why a genetic counselor or other healthcare professional can help you determine if you meet this criteria and may have an increased risk for Li-Fraumeni syndrome.
Treatment/Management for Li-Fraumeni syndrome
If you are tested and found to have a pathogenic variant in the TP53 gene, it is recommended to discuss your management plan with your healthcare team, and if available, to seek consultation through a specialized high-risk clinic. General recommendations are included here based on updated guidelines of the NCCN, but may be tailored to your specific medical and family history.
Breast cancer screening recommendations for women:
- Breast awareness starting at age 18
- Clinical breast exam every 6-12 months, starting at age 20-25, or at the age of earliest diagnosed breast cancer in the family if below age 20
- Breast screening:
- Age 20-29 (or at the age of earliest diagnosed breast cancer in the family if below age 20): annual breast MRI screening with contrast (preferred) or mammogram if MRI is unavailable
- Age 30-75: annual mammogram and breast MRI screening with contrast
- Over age 75: management should be considered on an individual basis
- For women with Li-Fraumeni who have been treated for breast cancer, screening of the remaining breast tissue with annual mammograms and breast MRI should continue
- Discuss option of risk-reducing mastectomy, including the psychosocial, social, and quality-of-life aspects of the procedure
To address the risk for other cancers:
- Annual comprehensive physical exam, including a neurologic exam, paying extra attention for signs or symptoms of rare cancers
- Therapeutic radiation treatment for cancer should be avoided when possible
- Consider colonoscopy screening every 2-5 years starting at age 25, or 5 years before the earliest known colon cancer in the family (whichever comes first)
- Annual dermatologic examination
- Annual whole body MRI; the brain may be examined as part of this or as a separate exam
- Additional surveillance based on family history of cancer
- Provide education regarding signs and symptom of cancer
Lynch syndrome, also called hereditary non-polyposis colorectal cancer (HNPCC) syndrome is caused by pathogenic (or harmful) variants in one of five known genes: MLH1, MSH2, MSH6, PMS2, and EPCAM. Seeing any of the following in a family could increase the chance for Lynch syndrome:
- Early-onset colorectal cancer (occurring before the age of 50 years)
- Early-onset uterine cancer (occuring before the age of 50 years)
- Multiple individuals diagnosed with colorectal and/or uterine cancer over several generations on the same side of the family
- One individual diagnosed with more than one type of cancer in their lifetime
- Patterns of stomach, small intestinal, ovarian, urinary tract, pancreatic, and glioma/glioblastoma (type of brain cancer), in addition to colorectal, or uterine cancer
Pathogenic variants that cause Lynch syndrome are inherited in an autosomal dominant pattern, meaning that children of someone with Lynch syndrome each have a 50% chance to also have Lynch syndrome and the related increased risks for cancer. Women and men both have the Lynch syndrome genes and have the same chances to inherit and pass down pathogenic variants in these genes, therefore both sides of the family are important to look at when trying to determine if someone has a higher chance to have Lynch syndrome.
It is important to remember that there are 5 genes that are known to cause Lynch syndrome and each of these genes have their own unique cancer risk numbers. The MLH1, MSH2, and EPCAM genes are associated with a higher risk of cancers (as described in the above table), while the MSH6 and PMS2 genes are still associated with an increased risk for cancer, just not as high as with MLH1, MSH2, and EPCAM. All individuals who have Lynch syndrome should be offered additional surveillance and screening regardless of which of the five genes is causing their Lynch syndrome.
Genetic Testing for Lynch Syndrome
Genetic testing for pathogenic variants that cause Lynch syndrome has been available for many years, and the testing methods have changed and improved over time. There are several different ways to approach to testing in these genes, depending on the history and any prior testing that may have been done. Different approaches include:
- Single site analysis: Testing specific to a known pathogenic variant in the family
- Full gene sequencing and rearrangement analysis: Comprehensive testing to search for all currently detectable variants in the genes.
- Gene panels: Newer, more broadly based gene tests that would include not only the Lynch syndrome genes, but other genes known or suspected to be associated with increased cancer risks
Immunohistochemistry (IHC) testing for Lynch Syndrome
This testing is different from other types of genetic tests because it is done specifically on a person’s cancer/tumor. Some hospitals complete this testing on all patients diagnosed with colon or uterine cancer as part of their treatment, while other facilities do not. The IHC test for Lynch syndrome (also called mismatch repair deficiency) is a screening test, meaning that it cannot tell us with 100% certainty whether or not someone has Lynch syndrome. It also may not find all patients that truly have Lynch syndrome. More commonly, positive results from IHC screening end up being a false positive, meaning that the person’s tumor looks like Lynch syndrome, but this is later ruled out by genetic testing. Individuals who have an abnormal IHC screening test should be referred to speak with a genetic counselor to discuss the results and help determine if genetic testing for Lynch syndrome is indicated and something that the patient wants to do. Many patients do not know if an IHC screening test was performed on their cancer, but you can ask your doctor about this information.
Who should be offered testing for Lynch Syndrome?
Other than someone’s personal history of cancer, another piece of information that we can use to help determine if someone is at a higher risk to have Lynch syndrome is your family history. The National Comprehensive Cancer Network (NCCN) is a group of medical professionals that regularly meet to look over any updates in research studies and determine recommendations for who should be considered at a higher risk for one of these pathogenic gene variants, and thus should be offered genetic testing.
Cancers that are associated with Lynch syndrome include: colon cancer, uterine cancer, ovarian cancer, pancreatic cancer, stomach cancer, small intestinal cancer, hepatobiliary cancer, brain cancer (specifically glioblastoma), urinary tract cancer (specifically renal pelvic type), and sebaceous skin tumors/cancer.
You should consider genetic counseling and possibly genetic testing if you see these cancers within your family history (make sure to include yourself in your family history if you have been diagnosed with cancer), specifically if you see:
- Colon cancer or uterine cancer under the age of 50
- An individual diagnosed with two separate colon cancers in their lifetime
- An individual diagnosed with two or more of the Lynch syndrome-associated cancers (listed above)
- An individual with colon cancer AND a close relative who has been diagnosed with a Lynch-syndrome associated cancer (listed above), with one of the cancers being diagnosed under the age of 50
- An individual with colon cancer AND two or more individuals within the same side of the family with a Lynch syndrome-associated (listed above) cancer at any age
These are some loose guidelines for who may be at an increased risk, but a medical professional, such as a genetic counselor, will be able to meet with you to further examine your family history and help determine if you meet criteria for genetic testing.
Treatment/Management for Lynch Syndrome
If you are tested and found to have Lynch syndrome, it is recommended to discuss your management plan with your healthcare team, and if available, to seek consultation through a specialized high-risk clinic. General recommendations are included here based on the updated guidelines of the NCCN, but may be tailored to your specific medical and family history.
All individuals with Lynch syndrome should have a colonoscopy every 1-2 years to look for colon polyps which may grow into colorectal cancer. This screening should start around age 20-25. The good news is that this screening for colon cancer is highly effective in reducing the chance that someone would develop colon cancer and reduces the severity of colon cancer.
An endoscopy (EGD) to look at the stomach and duodenum (the first part of the small intestine immediately beyond the stomach) may be considered for those who have a family history of stomach or small intestinal cancer or have other risk factors for these cancers (i.e. Asian descent).
Women at increased risk for uterine and ovarian cancer due to having Lynch syndrome are encouraged to consider prophylactic (preventative) removal of the uterus, ovaries, and fallopian tubes once they are certain they would not like any future pregnancies. For women who do not elect preventive surgery, screening for these cancers may include biopsies of the uterus, checking CA-125 blood levels to screen for ovarian cancer, and transvaginal ultrasound. However, these screenings have not proven to be effective and are not recommended by many medical organizations.
In families with Lynch syndrome and a history of pancreatic cancer, individualized screening may be offered based on the history, but no specific guidelines currently exist.
An annual urine test can be considered to screen for urinary tract cancers starting around age 30-35.
Individuals with Lynch syndrome should have regular physical exams with primary cancer physician including a neurological evaluation.
Neurofibromatosis, type 1 (NF1) is a genetic condition that can cause certain pigment (color) changes on the skin, an increased risk for certain types of cancer, and benign tumors in the skin, brain, and other parts of the body. The signs of NF1 can usually be found in childhood, and the first signs that are found are often the skin pigment changes, which can sometimes be very subtle. As people with NF1 get older, their chance to develop the tumors and cancer increase.
Individuals with NF1 can live normal lives, but they may develop serious health problems if they are not closely monitored by a healthcare provider who has experience with it. NF1 can potentially affect multiple body systems, and can affect individuals very differently. Individuals with NF1 are also at an increased risk to have scoliosis (curvature of the spine) or other bone abnormalities, as well as other tumors of the gastrointestinal tract or central nervous system. About half of people with NF1 also have learning disabilities.
Causes of NF1
We have over 20,000 different genes in the body. These genes are like instruction manuals for how to build a protein, and each protein has an important function that helps to keep our body working how it should. The NF1 gene makes a protein called neurofibromin. Neurofibromin works with other proteins to control how quickly the body’s cells multiply (called a tumor suppressor protein). If someone has a harmful change (called a pathogenic variant) in one of their NF1 genes, then their body does not make as much neurofibromin as it should. Without enough neurofibromin, there is not proper control over how fast our cells grow, which can lead them to multiply more cell growth than there should be. This unregulated cell growth can lead to tumors and the other signs and symptoms that we associate with NF1.
Pathogenic variants in the NF1 gene cause NF1, and are inherited in an autosomal dominant pattern, meaning that children of a carrier each have a 50% risk to inherit the pathogenic variant and associated health risks. Notably, women and men both have the NF1 genes and have the same chances to inherit and pass down variants in these genes. Therefore, both sides of the family are important when assessing inherited risk. About 50% (or 1 in 2 people) with NF1 will also have a parent with NF1. The other 50% have no family history and will be the first ones in their family with this condition.
It is estimated that approximately 1 in 3,000 people have NF1.
Genetic Testing for NF1
Genetic testing for NF1 is not always needed, as it can be diagnosed via physical exam by a genetics specialist. There are some instances where genetic testing can be helpful:
- If someone is suspected to have NF1 but this is not confirmed by the physical exam
- If a child has a suspicious tumor, and genetic testing would immediately impact their treatment
- If a parent who has NF1 would like to to do prenatal or preconception testing to determine if an embryo or pregnancy is also affected with NF1.
Genetic testing for pathogenic variants in NF1 is currently available, but there are a few different ways to approach testing:
- Single site analysis: Testing specific to a known pathogenic variant in the family
- Full gene sequencing and rearrangement analysis: Comprehensive testing to search for all currently detectable pathogenic variants in the gene
- Microarray analysis to detect large deletions in the NF1 gene
- Gene panels: Newer, more broadly based gene tests that would include not only the NF1 gene, but other genes known or suspected to be associated with the signs of NF1 or breast cancer. For example, pathogenic variants in the SPRED1 gene can result in skin findings that can be similar to NF1, but does not have the increased risk for tumors and cancer that individuals with NF1 have. The NF1 gene is also on some panels that are testing for an increased risk for breast cancer, along with genes such as BRCA1 and BRCA2.
Diagnosing NF1
NF1 is diagnosed by detailed physical examination (usually done by a geneticist) and family history. The geneticist can look for the signs of NF1 during the examination and usually can determine if someone has NF1 or not based on that physical exam.
NF1 is suspected in a person with any of the following:
- Six or more cafe-au-lait (light brown spots)
- Two or more neurofibromas (a type of benign nerve tumor), or one plexiform neurofibroma (a more specific type of neurofibroma); neurofibromas can range from small, discrete lumps to larger internal masses
- Freckles in the groin or underarm area
- Optic glioma (benign tumors on the nerve that connects the eyes to the brain, which can sometimes affect vision)
- Two or more Lisch nodules (benign growths, like freckles, on the iris of the eye)
- A first-degree relative (parent, sibling, or child) with NF1
- Bone abnormalities, such as sphenoid dysplasia (abnormal bone at the base of the skull) and tibial pseudarthrosis (when the larger of the two bones in the lower leg breaks and does not heal correctly)
Individuals with NF1 also have an increased risk for specific types of cancer, scoliosis, and learning delays. Because the diagnosis of NF1 is made by a doctor’s exam, genetic testing for NF1 is generally not recommended.
If you or your family meet any of the above criteria, a meeting with a pediatric or adult genetics specialist can be helpful to further determine if someone has NF1.
Associate Risks with NF1
The overall lifetime risk for cancer in NF1 is approximately 60%, which includes an increased risk for breast cancer, and certain gastrointestinal and central nervous system cancers (such as pheochromocytoma and malignant peripheral nerve sheath tumors). Studies have shown that women with NF1 have an elevated risk to develop breast cancer before the age of 50. After age 50, the risk to develop breast cancer returns to the level of other women in the general population.
Treatment/Management for NF1
Medical management for someone with NF1 and should be overseen by a medical provider who is familiar with NF1. Because NF1 affects people in very different ways, medical management should be specific to an individual’s needs. The frequency of each screening tool is tailored to an individual’s age. Some options for surveillance include:
- Regular eye exam
- Regular neurological exams and physicals
- Blood pressure monitoring
- MRI of the body and brain
- Breast cancer screening in women starting at age 30
The National Comprehensive Cancer Network (NCCN) recommends that women who have a diagnosis of NF1 should start annual mammogram screening at age 30, and should consider annual breast MRIs between the ages of 30 and 50. It is important to note the possibility that benign neurofibromas within the breast tissue may cause false-positive results on MRI. There is not currently any data or evidence to recommend risk-reducing mastectomies for women with NF1, but it may be considered on a case-by-case basis depending on family history. More personalized recommendations will take into account your personal and family history, and should be discussed with your medical provider.
Neurofibromatosis type 2 is a genetic predisposition to develop growths and tumors in different parts of the body. These slow growing tumors most commonly occur in the central nervous system, with the average age of onset in young adulthood (between 18-24 years old). Almost all people with NF2 will develop bilateral vestibular schwannomas (benign tumors that develop in the inner ear on both sides that can affect balance and hearing) by the age of 30. Complications from other central nervous system tumors can include changes in vision, weakness or numbness in the arms and legs, or fluid buildup in the brain. Some people with NF2 can also develop cataracts (a clouding of the lens of the eye) in either one or both of their eyes, and can occur as early as childhood.
Causes NF2
We have over 20,000 different genes in the body. These genes are like instruction manuals for how to build a protein, and each protein has an important function that helps to keep our body working how it should. The NF2 gene makes a protein called merlin. Merlin works with other proteins to control how quickly the body’s cells multiply (called a tumor suppressor protein). Merlin is made in the body’s central nervous system. If someone has a pathogenic (or harmful) variant in one of their NF2 genes, then their body does not make as much merlin as it should. Without enough merlin, there is not proper control over how fast our cells grow, which can lead them to multiply more cell growth than there should be. This unregulated cell growth can lead to tumors and the other signs and symptoms that we associate with NF2.
Pathogenic variants in the NF2 gene causes NF2, and are inherited in an autosomal dominant pattern, meaning that children of a carrier each have a 50% risk to inherit the pathogenic variant and associated health risks. Notably, women and men both have the NF2 genes and have the same chances to inherit and pass down variants in this gene. Therefore, both sides of the family are important when assessing inherited risk. About 50% (or 1 in 2 people) with NF2 will also have a parent with NF2. The other 50% have no family history and will be the first ones in their family with this condition.
It is estimated that approximately 1 in 33,000 people have NF2.
Genetic Testing for NF2
Genetic testing for pathogenic variants in NF2 is currently available, but there are a few different ways to approach testing:
- Single site analysis: Testing specific to a known pathogenic variant in the family
- Full gene sequencing and rearrangement analysis: Comprehensive testing to search for all currently detectable pathogenic variants in the gene
- Gene panels: Newer, more broadly based gene tests that would include not only the NF2 gene, but other genes known or suspected to be associated with these types of tumors.
Diagnosing NF2
A diagnosis of NF2 can be complex and therefore may be best conducted by a medical provider who is familiar with NF2. A diagnosis can usually be made based on a review of medical records (clinical diagnosis) and genetic testing for pathogenic variants in the NF2 gene.
NF2 is suspected in a person with any of the following:
- Bilateral vestibular schwannomas (formerly known as acoustic neuromas); benign tumors of the cranial nerves that control hearing and are associated with hearing loss, ringing of the ears, and trouble balancing
- Meningioma
- Neuropathy such as a facial palsy, foot drop or hand drop
- Schwannomas of the spine
- Hamartoma (benign tumor) of the retina
- Neurofibromas
If you or your family meet any of the above criteria, a meeting with a pediatric or adult genetics specialist can be helpful to further determine if someone has NF2.
Treatment/Management for NF2
Medical management for someone with NF2 should be overseen by a medical provider who is familiar with NF2 . Because NF2 affects people in very different ways, medical management should be specific to an individual’s needs. The frequency of each screening tool is tailored to an individual’s age. Some options for surveillance include:
- Regular neurological exams and physicals
- MRI of the brain
- Hearing evaluations
- Eye exams
More personalized recommendations will take into account your personal or family history, and should be discussed with your medical provider.
PTEN Hamartoma Tumor syndrome (PHTS) is a rare genetic condition that can affect children and adults. Over the years, many different names have been used to describe what we now consider the variability of features we see in PHTS. Other names for PHTS have included Cowden syndrome (CS), Bannayan-Riley-Ruvalcaba syndrome (BRRS), PTEN-related Proteus syndrome (PS), and Proteus-like syndrome.
Individuals with PHTS syndrome have an increased chance to develop benign growths, neurodevelopmental differences such as autism or intellectual disability and certain cancers in adulthood.
PHTS is caused by a harmful change (called a mutation or pathogenic variant) in the PTEN gene. PTEN gene variants are inherited in an autosomal dominant pattern, meaning that children someone who carries a variant each have a 50% risk to inherit the variant and associated risks. This also means women and men both have the PTEN gene and have the same chance to inherit and pass down variants in this gene. Therefore, both sides of the family are important when assessing inherited risk. However, the associated cancers and screening recommendations are different between men and women.
Cancers commonly associated with PHTS include:
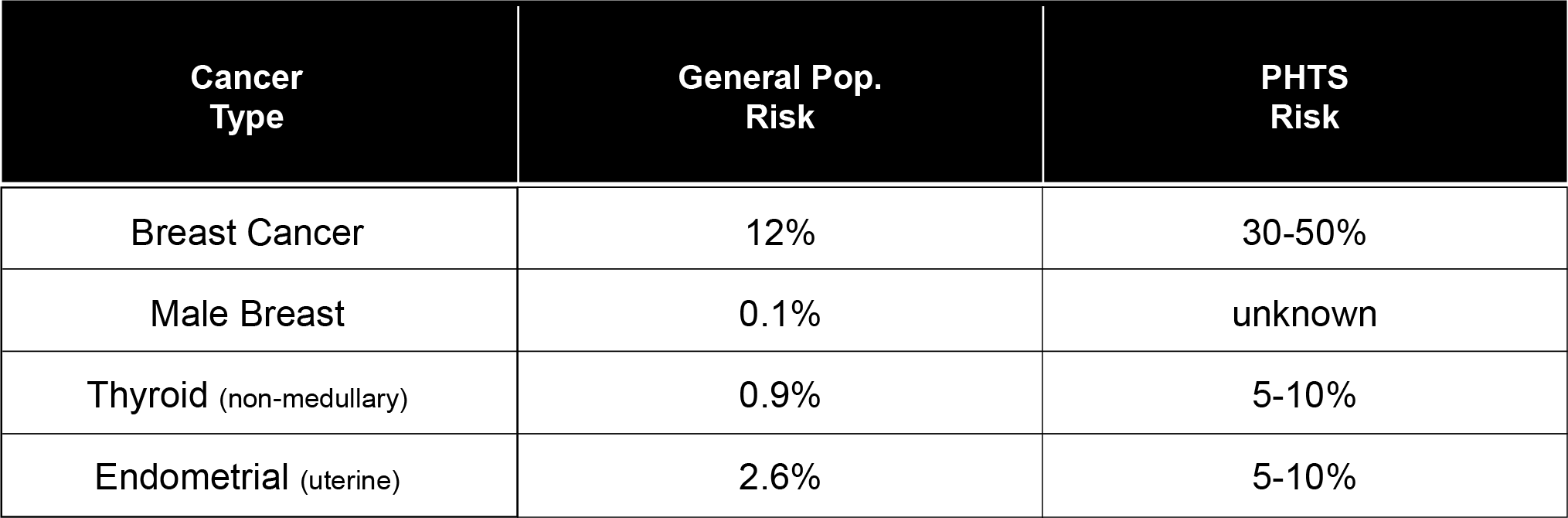
Some data suggests there may be a slight increased risk for other types of cancer, such as renal, glioblastoma, melanoma, lung, pancreatic, ovarian, and bladder cancers.
Some of the other characteristic signs of PTHS include:
- Larger than average head size (macrocephaly)
- Colon polyps (called hamartomas or ganglioneuromas)
- Benign skin growths (including trichilemmomas, keratoses, neuromas, papillomas, and lipomas)
- Pigmented spots (freckles) on the penis
- Thyroid goiters, adenomas, or nodules
- Lhermitte-Duclos (non-cancerous growth in the cerebellum)
Genetic Testing for PTEN
Genetic testing for pathogenic variants in PTEN are currently available, but there are a few different ways to approach testing:
- Single site analysis: Testing specific to a known pathogenic variant in the family
- Full gene sequencing and rearrangement analysis: Comprehensive testing to search for all currently detectable variants in the gene
- Gene panels: Newer, more broadly based gene tests that would include not only the PTEN gene, but other genes known or suspected to be associated with increased cancer risks
Determining whether an individual meets criteria for genetic testing for PTHS is very complex, and would likely be best left to a genetic counselor or other healthcare provider. PTHS can be diagnosed by a clinical exam performed by a physician familiar with PTHS, or by genetic testing for pathogenic variants in the PTEN gene.
Treatment/Management for PTEN
If you are tested and found to have a pathogenic variant in the PTEN gene, it is recommended to discuss your management plan with your healthcare team, and if available, to seek consultation through a specialized high-risk clinic. General recommendations are included here based on updated guidelines of the NCCN, but may be tailored to your specific medical and family history.
Childhood (below age 18):
- Yearly thyroid ultrasound starting at the time of diagnosis
- Yearly skin check with physical examination
- Consider neurodevelopmental evaluation
For Women:
- Breast awareness starting at age 18
- Clinical breast exam every 6-12 months starting at age 25, or 5-10 years before the earliest known breast cancer in the family (whichever comes first)
- Breast screening
- Annual mammogram and breast MRI screening with contrast starting at age 30-35, or 5-10 years before the earliest known breast cancer in the family (whichever comes first)
- For women over age 75, management should be considered on an individual basis
- Women who have been treated for breast cancer should still have screening of remaining breast tissue with annual mammogram and breast MRI.
- Encourage patient education and prompt response to symptoms of endometrial (uterine) cancer, such as abnormal bleeding. Consider annual random uterine biopsies and/or ultrasound beginning at age 30-35.
- Discussion of the option of a hysterectomy upon completion of childbearing; discuss degree of protection, extent of cancer risk, and psychosocial, social, and quality-of-life aspects.
- Discuss option of risk-reducing mastectomy, including degree of protection, extent of cancer risk, reconstruction options and psychosocial, social, and quality-of-life aspects.
For Men and Women:
- Annual comprehensive physical exam starting at age 18, or 5 years before the youngest age of diagnosis of cancer in the family (whichever comes first), with particular attention to the thyroid.
- Annual thyroid ultrasound starting at the time of diagnosis
- Colonoscopy starting at age 35 unless symptomatic, or if close relative with colon cancer before age 40, then start 5-10 years before the earliest known colon cancer in the family. Colonoscopy should be done every 5 years or more frequently if patient is symptomatic or polyps are found.
- Consider renal ultrasound starting at age 40, then every 1-2 years.
- Visits and procedures for skin lesions may necessitate visits with a dermatologist.
- Consider psychomotor assessment in children at diagnosis and brain MRI if there are symptoms.
- Education regarding the signs and symptoms of cancer.
Peutz-Jeghers syndrome is caused by inherited pathogenic (or harmful) variants in the STK11 gene. This condition is associated with increased risk for several types of cancer, as well as other characteristic features.
Pathogenic variants in STK11 are inherited in an autosomal dominant pattern, meaning that children someone who carries a pathogenic variant each have a 50% risk to inherit the variant and associated cancer risks. Notably, women and men both have the STK11 gene and have the same chances to inherit and pass down variants in this gene. Therefore, both sides of the family are important when assessing inherited risk. However, the associated cancers and lifetime risks are different between men and women.
The cancer risks in Peutz-Jeghers syndrome include:
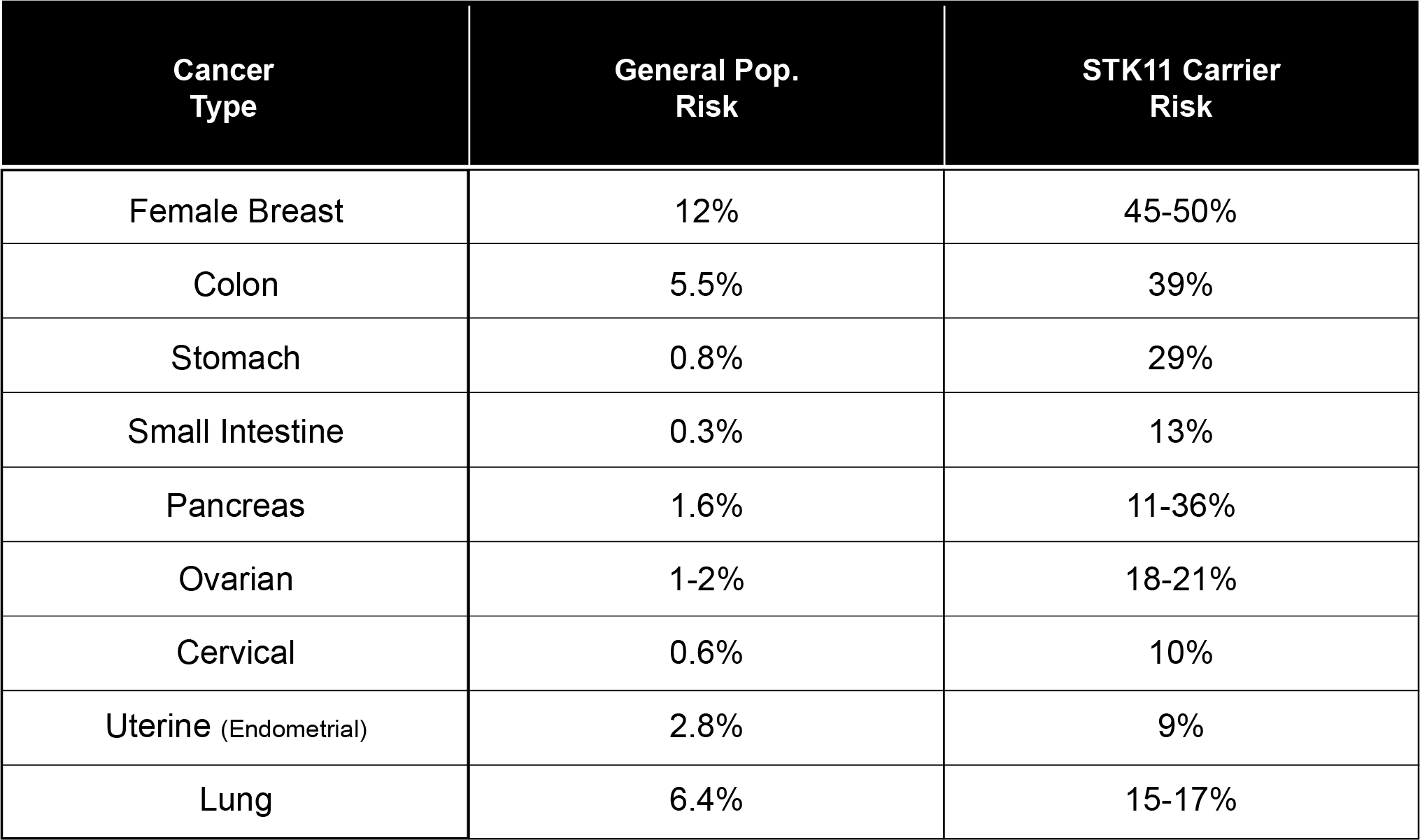
Other characteristics of Peutz-Jeghers syndrome include:
- Polyps (hamartomatous type) in the small intestine
- Freckles in unusual places (in/around the mouth, nose, eyes, fingers, groin)
Diagnosing Peutz-Jeghers Syndrome
Peutz-Jeghers syndrome is diagnosed if your personal and family history meets established clinical criteria (based on medical history, physical examination, and family history). Currently, the criteria for diagnosis, as determined by the National Comprehensive Cancer Network (NCCN), includes confirmation of two or more of the following in an individual:
- At least two Peutz-Jeghers-type hamartomatous polyps in the small intestine
- Mucocutaneous hyperpigmentation (freckles) of the mouth, lips, nose, eyes, genitals, or fingers
- Family history of Peutz-Jeghers syndrome
This condition can also be diagnosed by identification of a pathogenic variant in the STK11 gene through genetic testing.
Genetic Testing for Peutz-Jeghers Syndrome
A diagnosis of Peutz-Jeghers syndrome may also made by identification of a pathogenic variant in the STK11 gene. Genetic testing can be ordered with different approaches, depending on the history and available information:
- Single site analysis: Testing specific to a known pathogenic variant in the family
- Full gene sequencing and rearrangement analysis: Comprehensive testing to search for all currently detectable variants in the genes.
- Gene panels: Newer, more broadly based gene tests that would include not only the STK11 gene, but other genes known or suspected to be associated with increased cancer risks
Treatment/Management for Peutz-Jeghers syndrome
If genetic testing confirms a known pathogenic variant in the STK11 gene, it is recommended to discuss your management plan with your healthcare team, and if available, to seek consultation through a specialized high risk clinic. General recommendations are included here based on the updated guidelines of the (NCCN), but may be tailored to your specific medical and family history.
Breast cancer: starting at age 25, women should have annual mammograms and breast MRIs, and should have a clinical breast exam every 6 months.
- Colon cancer: starting in the late teens, men and women should have colonoscopy screening every 2-3 years
- Stomach cancer: starting in the late teens, men and women should have upper endoscopy screening every 2-3 years
- Small intestine cancer: starting at age 8-10, boys and girls should have small bowel screening (CT, MRI or video capsule endoscopy), with follow up by age 18 (or sooner if first screen found something), then repeat every 2-3 years
- Pancreatic cancer: starting at age 30-35, men and women should have MRI or endoscopic ultrasound every 1-2 years
- Ovarian, cervical, uterine cancers: starting at age 18-20, women should have annual pelvic exam and Pap smear; should also consider transvaginal ultrasound
- Testicular cancer: starting at age 10, boys should have annual testicular exam; should also observe for feminizing changes (e.g. breast growth)
- Lung cancer: Education on symptoms and smoking cessation
Tuberous sclerosis complex (TSC) is a genetic predisposition to growths in different parts of the body, and is caused by pathogenic (or harmful) variants in the TSC1 or TSC2 genes. Most often these growths are benign (non-cancerous) and occur on the skin, kidneys, heart, lung, and brain. Both men and women can be diagnosed with TSC. While signs of the condition can usually appear in childhood, some individuals do not know they have TSC until they are adults because they have mild symptoms, or because TSC is relatively uncommon.
Individuals with TSC can live normal lifespans, but they may develop serious health problems if they are not closely monitored by a healthcare provider experienced with the condition. TSC can potentially affect multiple body systems, however it affects individuals differently, even those within the same family.
Causes of Tuberous Sclerosis
Pathogenic variants in the TSC1 and TSC2 genes are inherited in an autosomal dominant pattern, meaning that children of someone who carries a pathogenic variant each have a 50% risk to inherit the variant and associated cancer risks. In one third (33%) of cases of TSC, someone has inherited a pathogenic variant in the TSC1 or TSC2 gene from a parent. In the other two thirds (66%) of cases, the variant was not inherited from a parent, and is something that is new to that person in the family (called de novo). Notably, women and men both have the TSC1 and TSC2 genes and have the same chances to inherit and pass down variants in these genes. Therefore, both sides of the family are important when assessing inherited risk.
There can be many different findings throughout many parts of the body in someone who has TSC. Here are some things that a doctor may look for to diagnose someone with TSC:
- Skin
- Multiple angiofibromas (skin colored, pink, or red bumps) of the face
- Multiple hypomelanotic macules (light colored spots)
- “Confetti” hypomelanotic macules
- Fibrous plaque on the forehead, cheeks, or scalp
- Shagreen patches (elevated area of orange peel-like skin) on the back, buttocks, or thighs
- Fibromas (benign tumor) around or under the fingernails or toenails
- Mouth
- Fibromas in the gums of the mouth
- Multiple pits in the tooth enamel
- Heart
- Cardiac rhabdomyoma (tumor of the heart in childhood)
- Brain
- Seizures
- Cortical tubers (benign lesions) found on a brain MRI
- Subependymal nodules (mass in the brain) found on brain MRI
- Subependymal giant cell astrocytoma (SEGA), a brain tumor that is noncancerous but can block of flow of cerebrospinal fluid in the brain (which is supposed to flow around you brain and down to you spinal cord)
- Eyes
- Hamartoma (benign flat lesion) of the retina or optic nerve
- Lung
- Lymphangioleiomyomatosis (LAM), which is when an unusual muscle cell invades the lungs, causing cysts and increasing risk for a collapsed lung. LAM mainly affects women
- Kidneys
- Angiomyolipomas (benign tumors) of the kidneys
- Fluid filled cysts
- Renal cell cancer (3% chance)
Genetic Testing for Tuberous Sclerosis
Genetic testing for pathogenic variants in TSC1 and TSC2 are currently available, but there are a few different ways to approach testing:
- Single site analysis: Testing specific to a known pathogenic variant in the family
- Full gene sequencing and rearrangement analysis: Comprehensive testing to search for all currently detectable variants in the gene
- Gene panels: Newer, more broadly based gene tests that would include not only the TSC1 and TSC2 genes, but other genes known or suspected to be associated with an increased risk for tumors or cancer.
Determining whether an individual meets criteria for genetic testing for TSC is very complex, and would likely be best left to a genetic counselor or other healthcare provider. TSC can be diagnosed by a clinical exam performed by a physician familiar with TSC, or by genetic testing for pathogenic variants in the TSC1 and TSC2 genes. About 95% of people who have a clinical diagnosis will be found to have a pathogenic variant in either the TSC1 or TSC2 genes. The remaining 5% may have a change in another gene that causes TSC that we have not yet discovered.
Treatment/Management for Tuberous Sclerosis
Medical management for someone with TSC can be complex, and should be overseen by a medical provider who is familiar with TSC. Because TSC affects people in very different ways, medical management should be tailored to an individual’s specific needs. Some options for treatment include:
- Medications to help treat seizures
- Surgery to remove problematic tumors
- Brain MRI every 1-3 years (more frequently if tumors are found)
- Abdominal MRI every 1-3 years
- Annual renal function assessment
- Annual dermatologic exam
- Dental exam every 6 months
This is not a comprehensive list of recommendations, as many of these are tailored to meet someone’s specific needs and current health concerns.
Von Hippel-Lindau (VHL) syndrome is a genetic predisposition to develop growths in different parts of the body, and is caused by pathogenic (or harmful) variants in the VHL gene. These growths may be benign (non-cancerous) or malignant (cancerous), and most commonly occur in the brain/central nervous system, kidneys, adrenal glands, reproductive organs, and pancreas. Both men and women can be diagnosed with VHL. While signs of VHL can appear in childhood and the teenage years, some individuals do not know they have it until they are older adults.
Individuals with VHL can live normal lives, but they may develop serious health problems if they are not closely monitored by a healthcare provider who has experience with it. VHL can potentially affect multiple body systems, and can affect individuals very differently.
Causes of Von Hippel Lindau
We have over 20,000 different genes in the body. These genes are like instruction manuals for how to build a protein, and each protein has an important function that helps to keep our body working how it should. The VHL gene makes a protein called the VHL protein. The VHL works with other proteins to form a complex (think of it like a vacuum) that picks up other proteins that are damaged or not needed any longer and gets rid of them. Getting rid of these extra proteins helps to make sure the cells can function how they are supposed to.
Pathogenic variants in the VHL gene are inherited in an autosomal dominant pattern, meaning that children of someone who has a pathogenic variant in the VHL gene have a 50% risk to inherit the variant and associated cancer risks. In 4 out of 5 (80%) cases of VHL, someone has inherited a variant in the VHL gene from a parent. In the other 1 in 5 (20%) cases, the variant was not inherited from a parent and is something that is new to that person in the family (called de novo). Notably, women and men both have the VHL gene and have the same chances to inherit and pass down variants in these genes. Therefore, both sides of the family are important when assessing inherited risk.
Genetic Testing for Von Hippel Lindau
Genetic testing for pathogenic variants in the VHL gene is currently available, but there are a few different ways to approach testing:
- Single site analysis: Testing specific to a known pathogenic variant in the family
- Full gene sequencing and rearrangement analysis: Comprehensive testing to search for all currently detectable variants in the gene
- Gene panels: Newer, more broadly based gene tests that would include not only the VHL gene, but other genes known or suspected to be associated with increased risk for tumors and cancer
A diagnosis of VHL can be complex and therefore may be best conducted by a clinician familiar with VHL. A diagnosis can be made based on a review of medical records (clinical diagnosis), or by genetic testing for mutations in the VHL gene. Nearly all people who have a clinical diagnosis will be found to have a mutation in the VHL gene upon testing.
VHL can be divided into two subgroups defined by the chance for particular types of tumors or cancer. Genetic testing can also help to distinguish these subtypes:
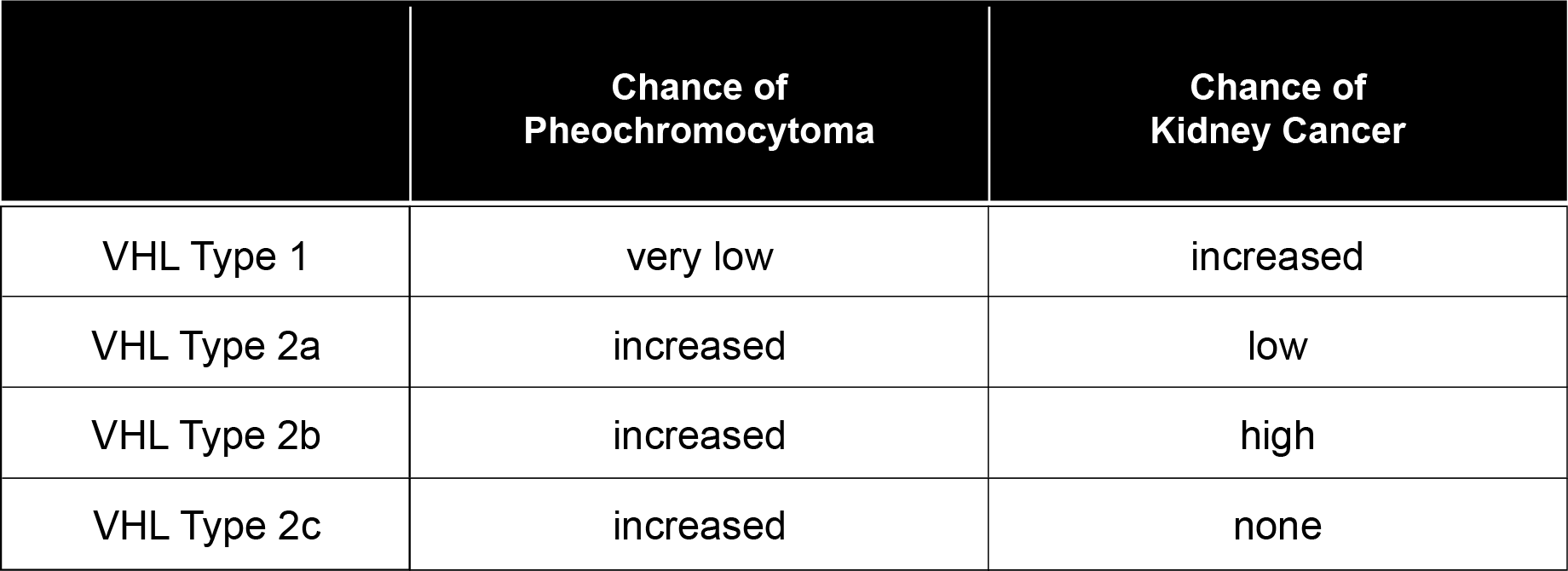
Associated Risks for Von Hippel Lindau
There can be many different findings throughout many parts of the body in someone who has VHL. Here are some things that a doctor may look for to diagnose someone with VHL:
- Central Nervous System
- Hemangioblastoma (a benign tumor that involves blood vessels) of the brain or spinal cord
- Auditory System
- Endolymphatic sac tumor (ELST; benign tumor of the inner ear)
- Eyes
- Hemangioblastoma of the retina (back of the eye)
- Kidneys
- Renal cell carcinoma (30 – 70% chance)
- Kidney cysts
- Adrenal Glands
- Pheochromocytoma (a benign tumor that causes elevated hormone levels)
- Paraganglioma (tumor similar to a pheochromocytoma that occurs outside of the adrenal glands)
- Pancreas
- Neuroendocrine tumor (PNET)
- Pancreatic cysts
- Reproductive Organs
- Broad ligament cystadenomas (benign tumor of the tissue that connects the female reproductive organs to the pelvis)
- Epididymal cystadenomas in males (benign tumor of the duct that carries sperm from the testis)
Treatment/Management for Von Hippel Lindau
Medical management for someone with VHL can be complex, and should be overseen by a medical provider who is familiar with VHL. Because VHL affects people in very different ways, medical management should be tailored to an individual’s specific needs. The frequency of each screening tool is tailored to an individual’s age. Some options for surveillance include:
- Eye exam and neurological exam
- Imaging of the abdomen using ultrasound and MRI
- Blood and urine testing for metanephrines
- Audiological (hearing) assessment
- MRI of the brain and spinal cord
This is not a comprehensive list of recommendations, as many of these are tailored to meet someone’s specific needs and current health concerns.
Wilms tumor is a type of kidney cancer that nearly always occurs in children, also called nephroblastoma.
Most Wilms tumors have no clear cause and no environmental risk factors have been identified either in childhood or during a woman’s pregnancy.
Causes of Wilms Tumor
Demographics
- Race: Kidney cancer is slightly more common in African Americans when compared with whites. Asian American children have the lowest rates of Wilms tumor.
- Gender: Wilms tumor is slightly more common in girls than boys
Medical History
Wilms tumor is more common in those children who have a history of other birth defects, such as:
- Hemihypertrophy (enlargement of limb or one half of the body)
- Aniridia (complete or partial lack of the iris of the eye)
- Undescended testes in boys
- Hypospadias (urethral opening in boys on the underside of the penis)
Family History
Wilms tumors can be diagnosed among multiple relatives within a family. It is thought that some of these patterns may be explained by a chromosomal or genetic change where a gene is missing.
Wilms tumor has been strongly associated with several genes and genetic syndromes
- WAGR syndrome is associated with aniridia (absence of the iris of the eye), genital and urinary abnormalities, and intellectual disability
- Beckwith-Wiedemann syndrome is associated with children who are larger than average, including their organs and specifically an enlarged tongue. One limb may be longer than the other.
- Denys-Drash syndrome results in kidney failure and reproductive organs fail to develop completely
- CTNNB1-related Wilms tumor
- AMER1-related Wilms tumor